Understanding the molecular basis of cancer initiation and progression is critical in developing effective treatment strategies. Mutations in genes encoding histone proteins that drive oncogenesis have been identified, converting these essential proteins into “oncohistones”. Understanding how oncohistone mutants, which are commonly single missense mutations, subvert the normal function of histones to drive oncogenesis requires defining the functional consequences of such changes. Histones genes are present in multiple copies in the human genome with 15 genes encoding histone H3 isoforms, the histone for which the majority of oncohistone variants have been analyzed thus far. With so many wildtype histone proteins being expressed simultaneously within the oncohistone, it can be difficult to decipher the precise mechanistic consequences of the mutant protein. In contrast to humans, budding and fission yeast contain only two or three histone H3 genes, respectively. Furthermore, yeast histones share ~90% sequence identity with human H3 protein. Its genetic simplicity and evolutionary conservation make yeast an excellent model for characterizing oncohistones.
- histone
- oncohistone
- budding yeast
- fission yeast
- epigenetics
- cancer
1. Introduction
2. Advantages of Using Yeast Models to Investigate Oncohistones
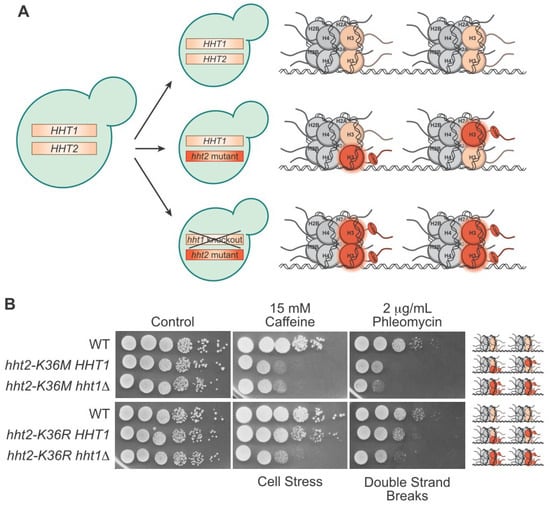
3. Progress towards Histone and Oncohistone Characterization via Yeast Models
This entry is adapted from the peer-reviewed paper 10.3390/jof9121187
References
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Mortality in the United States; 2021 NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2022; pp. 1–8.
- Hartwell, L.H. Yeast and cancer. Biosci. Rep. 2004, 24, 523–544.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231.
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253.
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482.
- Sankar, A.; Mohammad, F.; Sundaramurthy, A.K.; Wang, H.; Lerdrup, M.; Tatar, T.; Helin, K. Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat. Genet. 2022, 54, 754–760.
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001, 20, 5207–5218.
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell Biol. 2004, 24, 1884–1896.
- Spedale, G.; Timmers, H.T.; Pijnappel, W.W. ATAC-king the complexity of SAGA during evolution. Genes Dev. 2012, 26, 527–541.
- Liu, Y.; Zhang, Y.; Xue, H.; Cao, M.; Bai, G.; Mu, Z.; Yao, Y.; Sun, S.; Fang, D.; Huang, J. Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discov. 2021, 7, 32.
- Lachner, M.; Sengupta, R.; Schotta, G.; Jenuwein, T. Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 209–218.
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 540–545.
- Su, W.P.; Hsu, S.H.; Chia, L.C.; Lin, J.Y.; Chang, S.B.; Jiang, Z.D.; Lin, Y.J.; Shih, M.Y.; Chen, Y.C.; Chang, M.S.; et al. Combined Interactions of Plant Homeodomain and Chromodomain Regulate NuA4 Activity at DNA Double-Strand Breaks. Genetics 2016, 202, 77–92.
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018.
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640.
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113.
- Freitag, M. Histone Methylation by SET Domain Proteins in Fungi. Annu. Rev. Microbiol. 2017, 71, 413–439.
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48.
- Dai, J.; Hyland, E.M.; Yuan, D.S.; Huang, H.; Bader, J.S.; Boeke, J.D. Probing nucleosome function: A highly versatile library of synthetic histone H3 and H4 mutants. Cell 2008, 134, 1066–1078.
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009, 19, 674–681.
- Nakanishi, S.; Sanderson, B.W.; Delventhal, K.M.; Bradford, W.D.; Staehling-Hampton, K.; Shilatifard, A.A. comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 2008, 15, 881–888.
- Matsubara, K.; Sano, N.; Umehara, T.; Horikoshi, M. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells 2007, 12, 13–33.
- Collord, G.; Martincorena, I.; Young, M.D.; Foroni, L.; Bolli, N.; Stratton, M.R.; Vassiliou, G.S.; Campbell, P.J.; Behjati, S. Recurrent histone mutations in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2019, 184, 676–679.
- Kuranda, K.; Leberre, V.; Sokol, S.; Palamarczyk, G.; François, J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol. Microbiol. 2006, 61, 1147–1166.
- Chen, J.; Stubbe, J. Bleomycins: New methods will allow reinvestigation of old issues. Curr. Opin. Chem. Biol. 2004, 8, 175–181.
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol. 2021, 17, 403–411.
- Truong, D.M.; Boeke, J.D. Resetting the Yeast Epigenome with Human Nucleosomes. Cell 2017, 171, 1508–1519.
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell Biol. 1991, 11, 6317–6327.
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411.
- Georgakopoulos, T.; Thireos, G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J. 1992, 11, 4145–4152.
- Kuo, M.H.; Brownell, J.E.; Sobel, R.E.; Ranalli, T.A.; Cook, R.G.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 1996, 383, 269–272.
- Kruger, W.; Peterson, C.L.; Sil, A.; Coburn, C.; Arents, G.; Moudrianakis, E.N.; Herskowitz, I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995, 9, 2770–2779.
- Seol, J.H.; Kim, H.J.; Yoo, J.K.; Park, H.J.; Cho, E.J. Analysis of Saccharomyces cerevisiae histone H3 mutants reveals the role of the alphaN helix in nucleosome function. Biochem. Biophys. Res. Commun. 2008, 374, 543–548.
- Ngubo, M.; Reid, J.L.; Patterton, H.G. Distinct structural groups of histone H3 and H4 residues have divergent effects on chronological lifespan in Saccharomyces cerevisiae. PLoS ONE 2022, 17, e0268760.
- Yu, Y.; Srinivasan, M.; Nakanishi, S.; Leatherwood, J.; Shilatifard, A.; Sternglanz, R.A. conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol. Cell Biol. 2011, 31, 2311–2325.
- Lemon, L.D.; Kannan, S.; Mo, K.W.; Adams, M.; Choi, H.G.; Gulka, A.O.D.; Withers, E.S.; Nurelegne, H.T.; Gomez, V.; Ambrocio, R.E.; et al. A Saccharomyces cerevisiae model and screen to define the functional consequences of oncogenic histone missense mutations. G3 2022, 12, jkac120.
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell Biol. 2002, 22, 1298–1306.
- Smith, E.R.; Eisen, A.; Gu, W.; Sattah, M.; Pannuti, A.; Zhou, J.; Cook, R.G.; Lucchesi, J.C.; Allis, C.D. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc. Natl. Acad. Sci. USA 1998, 95, 3561–3565.
- Cooke, S.L.; Soares, B.L.; Müller, C.A.; Nieduszynski, C.A.; Bastos de Oliveira, F.M.; de Bruin, R.A.M. Tos4 mediates gene expression homeostasis through interaction with HDAC complexes independently of H3K56 acetylation. J. Biol. Chem. 2021, 296, 100533.
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861.
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife 2021, 10, e65369.
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 2017, 6, e27406.
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603.
- D’Arcy, S.; Luger, K. Understanding histone acetyltransferase Rtt109 structure and function: How many chaperones does it take? Curr. Opin. Struct. Biol. 2011, 21, 728–734.
- Bennett, R.L.; Bele, A.; Small, E.C.; Will, C.M.; Nabet, B.; Oyer, J.A.; Huang, X.; Ghosh, R.P.; Grzybowski, A.T.; Yu, T.; et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov. 2019, 9, 1438–1451.
- Shan, C.M.; Wang, J.; Xu, K.; Chen, H.; Yue, J.X.; Andrews, S.; Moresco, J.J.; Yates, J.R.; Nagy, P.L.; Tong, L.; et al. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife 2016, 5, e17903.
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849.
- Zhang, Y.; Shan, C.M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep. 2017, 7, 43906.