Billions of cells die in us every hour, and our tissues do not shrink because there is a natural regulation where Cell Death (CD) is balanced with cell division. The process in which cells eliminate themselves in a controlled manner is called Programmed Cell Death (PCD). The PCD plays an important role during embryonic development, in maintaining homeostasis of the body’s tissues, and in the elimination of damaged cells, under a wide range of physiological and developmental stimuli. Apoptosis is an RCD pathway that occurs inside eukaryotic cells and whose purpose is the death of the cell itself. Apoptosis is a “cellular suicide” in which a protein program of self-destruction triggered by extracellular or intracellular signals is set in motion. RCD means that the steps for cell degeneration are established, but that does not mean that the cell is predetermined to die; that is, there will be no apoptosis if there is no signal to initiate it. The role of apoptosis is important in many physiological and pathological processes of multicellular organisms, such as the morphogenesis of organs and tissues during embryonic development, in the maintenance and regeneration of tissues in the adult animal, in response to pathogens, or as a response to cellular stress and pathologies such as cancer. The number of cells that die by apoptosis is enormous, both during embryonic development and in the adult state, associated with caspases, that not only control apoptosis, but also proliferation, differentiation, cell form and cell migration.
- Programmed Cell Death
- caspase-dependent
- Anoikis
- Catastrophe Mitotic
- Pyroptosis
- Emperitosis
- Parthanatos
1. Anoikis
2. Mitotic Catastrophe
3. Pyroptosis
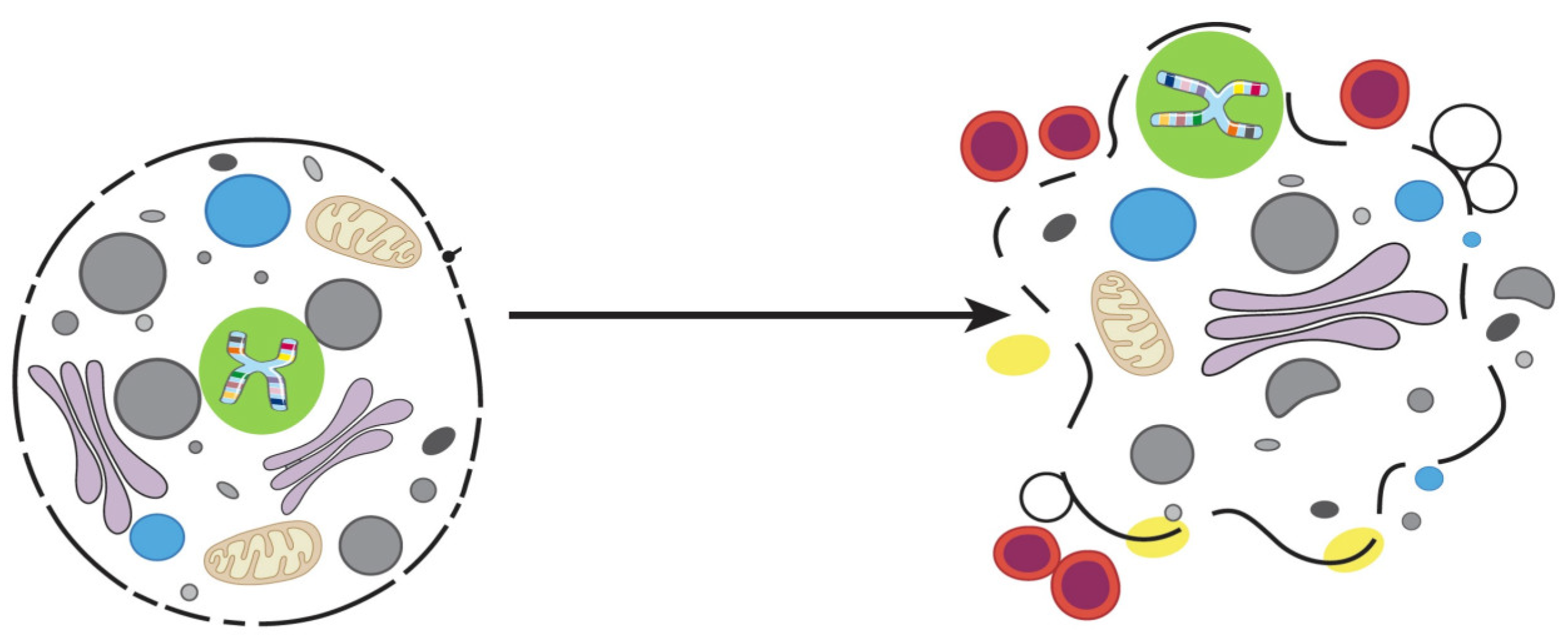
4. Emperitosis
5. Parthanatos
6. Cornification
This entry is adapted from the peer-reviewed paper 10.3390/proteomes12010003
References
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626.
- Woods, N.T.; Yamaguchi, H.; Lee, F.Y.; Bhalla, K.N.; Wang, H.G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 2007, 67, 10744–10752.
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740.
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562.
- Wang, J.; Luo, Z.; Lin, L.; Sui, X.; Yu, L.; Xu, C.; Zhang, R.; Zhao, Z.; Zhu, Q.; An, B.; et al. Anoikis-Associated Lung Cancer Metastasis: Mechanisms and Therapies. Cancers 2022, 14, 4791.
- Fernández-Lázaro, D.; Hernández, J.L.G.; García, A.C.; Martínez, A.C.; Mielgo-Ayuso, J.; Cruz-Hernández, J.J. Liquid Biopsy as Novel Tool in Precision Medicine: Origins, Properties, Identification and Clinical Perspective of Cancer’s Biomarkers. Diagnostics 2020, 10, 215.
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29.
- Portugal, J.; Mansilla, S.; Bataller, M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr. Pharm. Des. 2010, 16, 69–78.
- Li, K.; Wu, D.; Chen, X.; Zhang, T.; Zhang, L.; Yi, Y.; Miao, Z.; Jin, N.; Bi, X.; Wang, H.; et al. Current and emerging biomarkers of cell death in human disease. Biomed. Res. Int. 2014, 2014, 690103.
- Kimura, M.; Yoshioka, T.; Saio, M.; Banno, Y.; Nagaoka, H.; Okano, Y. Mitotic catastrophe and cell death induced by depletion of centrosomal proteins. Cell Death Dis. 2013, 4, e603.
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528.
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265.
- Niikura, Y.; Dixit, A.; Scott, R.; Perkins, G.; Kitagawa, K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J. Cell Biol. 2007, 178, 283–296.
- Huang, X.F.; Luo, S.K.; Xu, J.; Li, J.; Xu, D.R.; Wang, L.H.; Yan, M.; Wang, X.R.; Wan, X.B.; Zheng, F.M.; et al. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood 2008, 111, 2854–2865.
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162.
- Mu, R.; Wang, Y.B.; Wu, M.; Yang, Y.; Song, W.; Li, T.; Zhang, W.N.; Tan, B.; Li, A.L.; Wang, N.; et al. Depletion of pre-mRNA splicing factor Cdc5L inhibits mitotic progression and triggers mitotic catastrophe. Cell Death Dis. 2014, 5, e1151.
- Fernández-Lázaro, D.; Hernández, J.L.G.; García, A.C.; del Castillo, A.C.; Hueso, M.V.; Cruz-Hernández, J.J. Clinical Perspective and Translational Oncology of Liquid Biopsy. Diagnostics 2020, 10, 443.
- Zhang, Z.; Mao, W.; Wang, L.; Liu, M.; Zhang, W.; Wu, Y.; Zhang, J.; Mao, S.; Geng, J.; Yao, X. Depletion of CDC5L inhibits bladder cancer tumorigenesis. J. Cancer 2020, 11, 353–363.
- Monack, D.M.; Raupach, B.; Hromockyj, A.E.; Falkow, S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 9833–9838.
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114.
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214.
- LaRock, C.N.; Cookson, B.T. Burning down the house: Cellular actions during pyroptosis. PloS Pathog. 2013, 9, e1003793.
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142.
- Bortoluci, K.R.; Medzhitov, R. Control of infection by pyroptosis and autophagy: Role of TLR and NLR. Cell Mol. Life Sci. 2010, 67, 1643–1651.
- Salvesen, G.S. Dying from within: Granzyme B converts entosis to emperitosis. Cell Death Differ. 2014, 21, 3–4.
- Wang, S.; He, M.F.; Chen, Y.H.; Wang, M.Y.; Yu, X.M.; Bai, J.; Zhu, H.Y.; Wang, Y.Y.; Zhao, H.; Mei, Q.; et al. Rapid reuptake of granzyme B leads to emperitosis: An apoptotic cell-in-cell death of immune killer cells inside tumor cells. Cell Death Dis. 2013, 4, e856.
- Darmon, A.J.; Nicholson, D.W.; Bleackley, R.C. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 1995, 377, 446–448.
- Chinnaiyan, A.M.; Hanna, W.L.; Orth, K.; Duan, H.; Poirier, G.G.; Froelich, C.J.; Dixit, V.M. Cytotoxic T-cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr. Biol. 1996, 6, 897–899.
- Harraz, M.M.; Dawson, T.M.; Dawson, V.L. Advances in neuronal cell death 2007. Stroke 2008, 39, 286–288.
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009, 218, 193–202.
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. 2009, 14, 1116–1128.
- Jeggo, P.A. DNA repair: PARP—Another guardian angel? Curr. Biol. 1998, 8, R49–R51.
- Yu, S.W.; Andrabi, S.A.; Wang, H.; No, S.K.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319.
- Andrabi, S.A.; No, S.K.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313.
- Villanueva, R.; Ferreira, P.; Marcuello, C.; Usón, A.; Miramar, M.D.; Peleato, M.L.; Lostao, A.; Susin, S.A.; Medina, M. Key Residues Regulating the Reductase Activity of the Human Mitochondrial Apoptosis Inducing Factor. Biochemistry 2015, 54, 5175–5184.
- Novo, N.; Romero-Tamayo, S.; Marcuello, C.; Boneta, S.; Blasco-Machin, I.; Velázquez-Campoy, A.; Villanueva, R.; Moreno-Loshuertos, R.; Lostao, A.; Medina, M.; et al. Beyond a platform protein for the degradosome assembly: The Apoptosis-Inducing Factor as efficient nuclease involved in chromatinolysis. PNAS Nexus 2022, 2, pgac312.
- Fuchs, E.; Hanukoglu, I.; Marchuk, D.; Grace, M.P.; Kim, K.H. The nature and significance of differential keratin gene expression. Ann. N. Y. Acad. Sci. 1985, 455, 436–450.
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340.
- Elias, P.M. Stratum corneum defensive functions: An integrated view. J. Investig. Dermatol. 2005, 125, 183–200.
- Descargues, P.; Sil, A.K.; Karin, M. IKKalpha, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J. 2008, 27, 2639–2647.
- Wu, N.L.; Lee, T.A.; Tsai, T.L.; Lin, W.W. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J. Investig. Dermatol. 2011, 131, 874–883.
- Martens, M.D.; Karch, J.; Gordon, J.W. The molecular mosaic of regulated cell death in the cardiovascular system. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 868, 166297.
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta 2013, 1833, 3471–3480.