Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Influenza B virus (IBV) significantly impacts the health and the economy of the global population. WHO global health estimates project 1 billion flu cases annually, with 3 to 5 million resulting in severe disease and 0.3 to 0.5 million influenza-related deaths worldwide. Influenza B virus epidemics result in significant economic losses due to healthcare expenses, reduced workforce productivity, and strain on healthcare systems.
- influenza B virus
- treatment
- therapeutic options
- economic losses
1. Introduction
The influenza virus has a significant global impact, causing billions of cases, millions of severe cases, and hundreds of thousands of deaths annually [1]. Influenza is an acute respiratory infectious disease that is caused by the influenza virus. About 5% to 10% of adults and 20% to 30% of children are infected with the influenza virus every year [2]. Influenza viruses can be divided into three types, A, B, and C, according to the antigenicity of nucleoproteins, among which A and B are the primary pathogens that cause human influenza [3]. The frequency of localized epidemics caused by influenza B viruses has increased worldwide in recent years [4][5]. Influenza viruses rapidly evolve with antigenic diversity, resulting in new hemagglutinin and neuraminidase proteins. Antigenic shifts can cause pandemics, and antigenic mutation contributes to global impact [4]. Vaccinations since the 1960s have reduced the number of influenza cases, and antiviral drugs (neuraminidase and viral polymerase inhibitors) treat influenza B. The vaccine’s efficacy is approximately 60% in a good season. In case of mismatch with current circulating strains, its effectiveness decreases from 10% to 20% [6]. However, the vaccine’s estimated overall efficacy stands at 38% (62% against the flu A[H1N1] pdm09 viruses, 22% against flu A[H3N2] viruses, and 50% against the influenza b virus) [7]. Several factors influence the efficiency of influenza vaccines [7]. One of the major factors is that the genes in various strains of influenza viruses undergo constant mutations, altering the surface proteins and enabling the virus to evade the host’s immunological response. This phenomenon, termed antigenic variation, allows the virus to avoid detection, thereby increasing its virulence by circumventing the body’s pre-existing immune defense. In cases where the mutation is gradual and minor, the host (human) may manage to trigger an immunological response by producing antibodies. This gradual transformation over time is called antigenic drift [8]. Other factors influencing vaccine efficacy include the recipient’s age and health, the types and subtypes of circulating virus strains, and the degree of homology between the circulating virus and those included in the vaccine [7]. However, antiviral drug resistance is a serious concern.
2. Epidemiological Histories
This section summarizes the historical background of influenza virus circulation and its impact on epidemiology, immunology, and vaccination. Since 1889, several Influenza A virus pandemics have significantly affected the human immune system, with the H3N8 and H1N1 viruses presumed responsible for the 1889 and 1918 pandemics (Figure 1) [9].
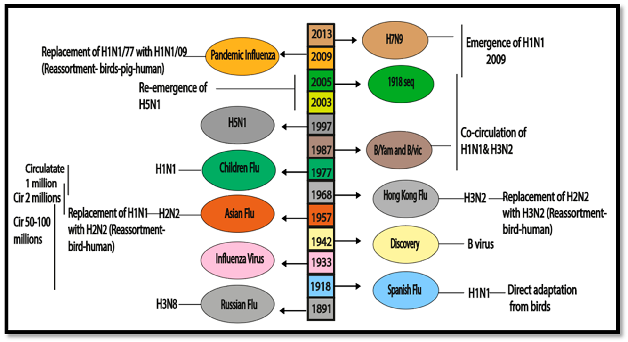
Figure 1. Epidemiological history and co-circulation of influenza virus.
The H1N1 virus, causing the deadliest pandemic in modern history with 50 to 100 million deaths worldwide, was identified through sequencing in 1997 and revived in 2005 [10][11]. Following the 1918 pandemic, H1N1 viruses continued circulating in humans, and the 1957 Asian pandemic introduced the H2N2 virus, causing approximately 2 million deaths [12]. The emergence of the H3N2 virus during the Hong Kong pandemic in 1968 led to the replacement of the H2N2 subtype due to population resistance to sub-immuno-dominant components [13]. In 1977, the H1N1 virus re-emerged, co-circulating with H3N2 until the sixth pandemic in 2009, caused by the introduction of a unique H1N1 lineage [14]. Zoonotic strains, such as H5N1, H6N2, H7N1, H7N7, and H9N2, have been reported to infect humans [15]. In 2013, H7N9 posed a serious threat to human health in China [16]. Influenza B viruses have been co-circulating since 1891, leading to a complex immunological environment. The WHO identified B/Yamagata/16/1988 (B/Yamagata lineage) and B/Victoria/2/1987 (B/Victoria lineage) by monitoring in the late 1980s [17].
The influenza virus pandemic also leads to economic loss. The present study concluded a short comparative analysis between China and the USA. Both China and the USA incur substantial economic losses due to influenza. China’s larger population and higher population density can lead to rapid transmission and significant healthcare costs and absenteeism [18]. The lower population density does not prevent substantial losses in the USA, with higher per capita healthcare costs and productivity impacts. Factors such as vaccination rates, healthcare infrastructure, and public health policies influence the extent of these economic burdens in each country [19].
3. Organization of Genome
IBVs are from the Orthomyxoviruses family, which comprises negative-sense single-stranded RNA with a segmented genome. IBVs include eight segments and are encodes for glycoproteins, matrix proteins, BM2 ion channels, polymerase proteins, nuclear export proteins, nonstructural proteins, and nucleoproteins (Figure 2) [20][21]. None of the IAV “accessory” proteins, such as PB1-F2 or PA-X, are encoded by IBVs. In general, there are significant differences between IAV and IBV proteins in terms of the length, function, and composition of amino acids [22]. IBV proteins are discussed below.
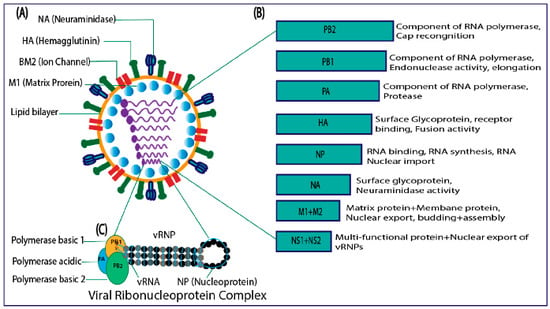
Figure 2. (A) Structure of influenza B virus. (B) Genome organization. (C) viral ribonucleoprotein complex.
3.1. The Hemagglutinin
IBV’s HA protein facilitates virus attachment to host cells through interactions with sialic acids [23]. It can bind to both α-2,3- and α-2,6-linked sialic acids. Structurally, IBV HA forms a functional homo-trimer with a globular membrane-distal region and an extended membrane-proximal region. Notably, there are significant differences in the receptor-binding site (RBS) between IBV HA and IAV HA, particularly at position 95, where IBV has phenylalanine, while IAV has tyrosine [17]. Phenylalanine at position 95 in IBV HA contributes to its lower affinity and ability to bind human-like and avian-like receptor analogues. Mutations that replace phenylalanine with tyrosine improve receptor binding similar to IAV and reduce adherence to avian-like receptor analogues [21]. Unlike IAV, the HA protein of IBV does not play a significant role in pathogenicity [20].
3.2. The Neuraminidase
IBV’s NA protein, like IAV NA, cleaves sialic acid to release new virions from the cell surface. Despite a 30% sequence homology, IBV NA has structural similarities to IAV NA. IBV NA forms a tetramer with a Ca2+-binding region and two glycosylation regions (Asn143 and Asn283) in each monomer. Asn143 is conserved across influenza viruses, while Asn283 is specific to IBV [22]. The active site of NA consists of 19 conserved residues, including R118, D151, R152, R224, E276, R292, R371, and Y406, which interact with sialic acids, and E119, R156, W178, S179, D198, I222, E227, H274, E277, N294, and E425, which maintain its structural integrity. Neuraminidase inhibitors (NAIs) approved for treating influenza may not effectively target the IBV [23].
3.3. The NB Protein
IBV’s segment 6 encodes NB, overlapping with the NA gene. NA’s start codon follows NB’s start codon by four nucleotides [20]. NB is a glycoprotein of around 100 amino acids in the virion, with modified N-glycosylation regions containing N-acetyl-glucosamine residues [20]. The palmitoylation of C-terminal residues is required for NB’s cell surface transport [24]. Previous studies suggest NB’s potential ion channel functionality based on comparisons with influenza A virus’s M2 protein [25]. Assessing NB’s electrophysiological properties using lipid bilayers is challenging due to hydrophobic proteins and peptides exhibiting channel-like behavior [26]. Studies questioning NB’s channel activity contradict its assumed role as an ion channel protein, despite amantadine’s inability to inhibit influenza B virus growth [20]. NB’s purpose and function have remained enigmatic for over 30 years, and understanding it may contribute to the development of innovative live attenuated vaccines [27].
3.4. The Matrix Protein (BM1)
IBV’s M1 protein, encoded in segment 7, is vital in the B/Ann Arbor/1/66 master donor strain of the live attenuated influenza vaccine (LAIV) and IBV transformation in mice [28]. The precise role of M1 in the IBV life cycle is unclear, but it shuttles between the nucleus and cytoplasm, possessing nuclear export signals and a nuclear localization signal (NLS) [29]. Further research is required to elucidate the specific functions of BM1 and its resemblance to IAV M1.
3.5. The BM2 Ion Channel
BM2, encoded in segment 7, enters virions via the trans-Golgi network (TGN). It integrates vRNP similar to AM2, supporting virus viability. BM2 exhibits ion-conducting activity and shares similarities with AM2 in endosome acidification and Golgi pH adjustment. Its crystal structure reveals coiled-coil tetramers with polar-pore-lining residues, rendering adamantanes ineffective against BM2. Recent studies suggest a potential role of BM2 in inhibiting p53-mediated apoptosis and transcription. BM2 is expressed from a single mRNA molecule that translates both BM2 and M1 coding sequences with overlapping termination and start codons. The 45 nucleotides preceding the pentanucleotide sequence (UAAUG) within the M1-coding region are critical for BM2 expression. The upstream sequence of the pentanucleotide shows homology with the small ribosomal subunit (18S component), and translation initiation factor eIF3 and the mRNA-rRNA association are essential for BM2 expression. Initially discovered in BM2, this termination–reinitiation process is observed in other viruses, including caliciviruses [30][31][32][33][34].
3.6. The Nucleocapsid Protein
IBV nucleoprotein (BNP) differs significantly from IAV nucleoprotein (ANP) in structure and function. BNP lacks ANP’s NLSs and has a longer N-terminus with a unique 50 amino acid region. BNP’s structure includes a head, body, and long tail, with essential lysine clusters for RNA binding in the charged loop (residues 125–149). BNP’s extensible tail loop facilitates homo-oligomer formation, similar to ANP [22][35]. A conserved salt bridge (R472-E395 in BNP and R416-E339 in ANP) stabilizes loops and promotes homo-oligomerization, making it a potential therapeutic target [36]. BNP’s N-terminus is involved in nuclear localization through the K44RTR47 motif, and its removal affects virus replication and transcription [37].
3.7. The NS1 Protein
IBV’s NS1 protein (segment 8) is vital for virus replication [38]. It has an N-terminus (residues 1–90), C-terminus (residues 120–281), and associated domain (residues 91–119) [39]. NS1B localizes in the cytoplasm and interacts with importin α3, SC35 nuclear factor, and RNA. Unlike IAVs, NS1B does not hinder mRNA export but affects antiviral and host mRNA export [40]. NS1B inhibits IFN-αβ activation and nuclear translocation, increasing IFN expression and promoter activation in IBV when absent [41]. NS1B acts as an IFN response inhibitor, complementing NS1A IAV growth [42]. The C-terminus inhibits IFN promoter activity, while the N-terminus limits PKR kinase activation. NS1B blocks interferon-stimulated ISG15 protein, contributing to IBV host restriction. NS1B and NS1A have distinct roles in evading the innate immune response, with ISG15 playing a critical antiviral role against IBV. NS1B-deleted viruses attenuate in IFN-insufficient Vero cells, indicating additional functions of NS1B [43].
3.8. The NEP or NS2 Protein
IBV’s NEP facilitates nuclear export with a specific signal sequence in its N-terminus. It accumulates in the nucleus during late infection and then relocates to the cytoplasm near the plasma membrane for virion budding. NEP is a component of the vRNP complex in virions, and in contrast to IAV, NEP directly interacts with vRNP and BM1 in IBV. While the exact role of IBV NEP remains uncertain, it likely shares a similar function to its IAV counterpart [22][44].
3.9. Noncoding Sequences
Influenza viruses have noncoding regions that contain packaging signals and act as viral promoters for replication. These regions are crucial for vRNA replication in IBV [45]. Highly conserved sequences near the termini, such as 5′-AGCAGAAGC-3′ and 3′-TCATCxTTGT-5′, form secondary RNA structures through complementarity. Noncoding sequences, varying between segments but consistent within each segment, are located alongside the coding sequences in the long terminal regions. Understanding these noncoding sequences is essential for reverse genetics systems and creating chimeric IAVs/IBVs for potential vaccines [5][10][46].
4. Life Cycle of Influenza Virus
The influenza virus life cycle begins with HA binding to sialyl oligosaccharides on the host cell surface, leading to viral attachment. The virus enters the host cell through receptor-mediated endocytosis and undergoes fusion with the endosomal membrane, triggered by a pH change and the M2 ion channel. This releases uncoated vRNP complexes into the cytosol. Once inside the nucleus, vRNP complexes replicate and transcribe vRNA, generating viral proteins. The newly produced vRNAs are transported to the cell membrane, cleaved by neuraminidase, and released as influenza, of which a virion is shown in Figure 3 [10].
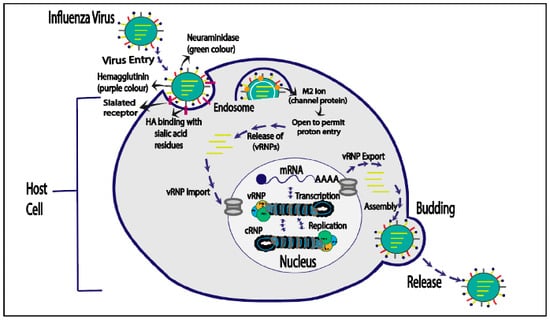
Figure 3. Life cycle of influenza virus.
4.1. Influenza Virus Entry
The influenza virus life cycle initiates with viral attachment to the host cell surface, facilitated by the interaction of HA with sialyloligosaccharides on the cell membrane. Using receptor-mediated endocytosis, the virus enters the host cell, where it is contained inside an endosome. The low pH in late endosomes triggers a structural change in HA, controlled by the M2 ion channel. The process culminates in a fusion of the viral and endosomal membranes. This results in the release of uncoated viral ribonucleoprotein (vRNP) complexes into the host cell cytoplasm. Neuraminidase cleaves freshly generated vRNPs after they have been transported to the cytoplasm and integrated by viral proteins at budding sites within the host cell membrane. These vRNPs are then discharged as influenza virions. (Figure 3) [47].
4.2. Viral Ribonucleoproteins’ (vRNPs) Entry into the Cell Nucleus
The viral RdRp is composed of three different types of viral proteins (PA, PB1, and PB2). The nuclear localization signals (NLSs) of these proteins have the potential to bind to the cellular nuclear import apparatus, which makes it easier for the proteins to enter the nucleus. The most important NLS for the vRNP nuclear entrance is still unknown. Notably, the nuclear import happens via a Crm1-dependent pathway, which involves binding several karyopherins like importin α and β [48].
4.3. Replication and Transcription
The influenza virus, with negative-sense RNA strands, undergoes transcription by converting the genome into positive-standard RNA, acting as a template for the synthesis of viral RNA. Genome replication occurs without primers as the virus’s RNA-dependent RNA polymerase (RdRp) begins internal synthesis. Partial inverse complementarity at the genome’s extreme ends facilitates the formation of corkscrew structures. The complete replication mechanism remains unknown despite the presence of di-nucleotide base pairs [49]. Mature cellular messenger RNAs (mRNAs) have a poly(A) tail and a 5′ methylated cap. Although vRNPs lack 5′ caps, they possess poly(A) tails. The absence of a 5′ cap in the viral genome puzzled researchers until they discovered that the viral mRNAs had a poly(A) tail and a 5′ methylation cap. Further research revealed that the 5′ methylation caps of viral mRNAs are associated with cellular mRNAs, leading to the “cap-snatching” method [50]. The viral RdRp, composed of PA, PB1 and PB2, utilizes PB2 to cleave the 3′ caps’ regions with 10–15 nucleotides. The viral RdRp initiates transcription using cellularly bound RNA fragments [51]. The cellular cap synthesis complex activates when transcription initiation involves cellular RNA polymerase II (Pol II) attaching to DNA, with the phosphorylation of serine five on Pol II’s C-terminal repeat domain (CTD). Research indicates that the influenza RdRp prefers binding to this form of Pol II, suggesting a potential site for “cap snatching” [52].
4.4. vRNPs Export
vRNPs most likely use the CRM1-dependent method to exit the nucleus through nuclear pores. In the lack of clear GTP hydrolysis activity, direct contacts between NP and CRM1 suggest an unusual export mechanism. M1’s C-terminal end directly binds to vRNPs, and its N-terminal region has a putative NLS connected to vRNP import. M1’s N-terminal segment can bind to NEP, hiding the NLS. NEP forms a compound with CRM1 after GTP hydrolysis, which may indicate a “daisy-chain” export route for vRNPs from the nucleus [52].
4.5. Budding Host Cell Plasma Membrane
After the exit of vRNPs from the nucleus, the virus assembles and leaves the host cell by utilizing the plasma membrane. Essential viral proteins (HA, NA, and M2) are crucial for particle generation. Elongated particles result from modifying the M2 tail. M1, beneath the lipid bilayer, aids in particle closure and budding. Sialic acid cleavage by NA is essential for viral particle release from the plasma membrane [47][53][54].
This entry is adapted from the peer-reviewed paper 10.3390/cimb46010014
References
- Peasah, S.K.; Azziz-Baumgartner, E.; Breese, J.; Meltzer, M.I.; Widdowson, M.-A. Influenza cost and cost-effectiveness studies globally—A review. Vaccine 2013, 31, 5339–5348.
- Fischer, W.A., II; Gong, M.; Bhagwanjee, S.; Sevransky, J. Global Burden of Influenza as a Cause of Cardiopulmonary Morbidity and Mortality. Glob. Heart 2014, 9, 325–336.
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.R.; Ebrahimpour, S. A brief review of influenza virus infection. J. Med. Virol. 2021, 93, 4638–4646.
- Niederman, M.S.; Krilov, L.R. Acute lower respiratory infection in developing countries. Lancet 2013, 381, 1341–1342.
- Nguyen, Y.T.; Graitcer, S.B.; Nguyen, T.H.; Tran, D.N.; Pham, T.D.; Le, M.T.; Tran, H.N.; Bui, C.T.; Dang, D.T.; Nguyen, L.T.; et al. National surveillance for influenza and influenza-like illness in Vietnam, 2006–2010. Vaccine 2013, 31, 4368–4374.
- Eisenstein, M. Towards a universal flu vaccine. Nature 2019, 573, S50–S52.
- Grohskopf, L.A. Prevention and control of seasonal influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2019–2020 influenza season. MMWR. Recomm. Rep. 2019, 68, 1–21.
- Nypaver, C.; Dehlinger, C.; Carter, C. Influenza and influenza vaccine: A review. J. Midwifery Women’s Health 2021, 66, 45–53.
- Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the future for influenza preimmunity—Looking back at influenza virus history to infer the outcome of future infections. Viruses 2019, 11, 122.
- Tumpey, T.M.; Basler, C.F.; Aguilar, P.V.; Zeng, H.; Solórzano, A.; Swayne, D.E.; Cox, N.J.; Katz, J.M.; Taubenberger, J.K.; Palese, P.; et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 2005, 310, 77–80.
- Zimmer, S.M.; Burke, D.S. Historical perspective—Emergence of influenza A (H1N1) viruses. N. Engl. J. Med. 2009, 361, 279–285.
- Nelson, M.I.; Viboud, C.; Simonsen, L.; Bennett, R.T.; Griesemer, S.B.; St George, K.; Taylor, J.; Spiro, D.J.; Sengamalay, N.A.; Ghedin, E.; et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog. 2008, 4, e1000012.
- Jester, B.J.; Uyeki, T.M.; Jernigan, D.B. Fifty years of influenza A (H3N2) following the pandemic of 1968. Am. J. Public Health 2020, 110, 669–676.
- Broadbent, A.J.; Subbarao, K. Influenza virus vaccines: Lessons from the 2009 H1N1 pandemic. Curr. Opin. Virol. 2011, 1, 254–262.
- Hannoun, C. The evolving history of influenza viruses and influenza vaccines. Expert Rev. Vaccines 2013, 12, 1085–1094.
- Koutsakos, M.; Nguyen, T.H.; Barclay, W.S.; Kedzierska, K. Knowns and unknowns of influenza B viruses. Future Microbiol. 2016, 11, 119–135.
- Wang, Q.; Tian, X.; Chen, X.; Ma, J. Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc. Natl. Acad. Sci. USA 2007, 104, 16874–16879.
- Gong, H.; Shen, X.; Yan, H.; Lu, W.Y.; Zhong, G.J.; Dong, K.G.; Yang, J.; Yu, H.J. Estimating the disease burden of seasonal influenza in China, 2006–2019. Zhonghua Yi Xue Za Zhi 2021, 101, 560–567.
- Dorratoltaj, N.; Marathe, A.; Lewis, B.L.; Swarup, S.; Eubank, S.G.; Abbas, K.M. Epidemiological and economic impact of pandemic influenza in Chicago: Priorities for vaccine interventions. PLoS Comput. Biol. 2017, 13, e1005521.
- Ni, F.; Mbawuike, I.N.; Kondrashkina, E.; Wang, Q. The roles of hemagglutinin Phe-95 in receptor binding and pathogenicity of influenza B virus. Virology 2014, 450, 71–83.
- Burmeister, W.; Ruigrok, R.; Cusack, S. The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 1992, 11, 49–56.
- Burnham, A.J.; Baranovich, T.; Govorkova, E.A. Neuraminidase inhibitors for influenza B virus infection: Efficacy and resistance. Antivir. Res. 2013, 100, 520–534.
- Demers, A.; Ran, Z.; Deng, Q.; Wang, D.; Edman, B.; Lu, W.; Li, F. Palmitoylation is required for intracellular trafficking of influenza B virus NB protein and efficient influenza B virus growth in vitro. J. Gen. Virol. 2014, 95 Pt 6, 1211.
- Fischer, W.B.; Pitkeathly, M.; Wallace, B.A.; Forrest, L.R.; Smith, G.R.; Sansom, M.S.P. Transmembrane peptide NB of influenza B: A simulation, structure, and conductance study. Biochemistry 2000, 39, 12708–12716.
- Tosteson, M.T.; Auld, D.S.; Tosteson, D.C. Voltage-gated channels formed in lipid bilayers by a positively charged segment of the Na-channel polypeptide. Proc. Natl. Acad. Sci. USA 1989, 86, 707–710.
- Hatta, M.; Kawaoka, Y. The NB protein of influenza B virus is not necessary for virus replication in vitro. J. Virol. 2003, 77, 6050–6054.
- McCullers, J.A.; Hoffmann, E.; Huber, V.C.; Nickerson, A.D. A single amino acid change in the C-terminal domain of the matrix protein M1 of influenza B virus confers mouse adaptation and virulence. Virology 2005, 336, 318–326.
- Cao, S.; Jiang, J.; Li, J.; Li, Y.; Yang, L.; Wang, S.; Yan, J.; Gao, G.F.; Liu, W. Characterization of the nucleocytoplasmic shuttle of the matrix protein of influenza B virus. J. Virol. 2014, 88, 7464–7473.
- Wang, J.; Pielak, R.M.; McClintock, M.A.; Chou, J.J. Solution structure and functional analysis of the influenza B proton channel. Nat. Struct. Mol. Biol. 2009, 16, 1267–1271.
- Mould, J.A.; Paterson, R.G.; Takeda, M.; Ohigashi, Y.; Venkataraman, P.; Lamb, R.A.; Pinto, L.H. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev. Cell 2003, 5, 175–184.
- Zhang, H.; Yu, H.; Wang, J.; Zhang, M.; Wang, X.; Ahmad, W.; Duan, M.; Guan, Z. The BM2 protein of influenza B virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Mol. Cell. Biochem. 2015, 403, 187–197.
- Horvath, C.M.; Williams, M.A.; Lamb, R.A. Eukaryotic coupled translation of tandem cistrons: Identification of the influenza B virus BM2 polypeptide. EMBO J. 1990, 9, 2639–2647.
- Hatta, M.; Kohlmeier, C.K.; Hatta, Y.; Ozawa, M.; Kawaoka, Y. Region required for protein expression from the stop-start pentanucleotide in the M gene of influenza B virus. J. Virol. 2009, 83, 5939–5942.
- Powell, M.L.; Napthine, S.; Jackson, R.J.; Brierley, I.; Brown, T.D.K. Characterization of the termination–reinitiation strategy employed in the expression of influenza B virus BM2 protein. RNA 2008, 14, 2394–2406.
- Ng, A.K.-L.; Lam, M.K.-H.; Zhang, H.; Liu, J.; Au, S.W.-N.; Chan, P.K.-S.; Wang, J.; Shaw, P.-C. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J. Virol. 2012, 86, 6758–6767.
- Shen, Y.-F.; Chen, Y.-H.; Chu, S.-Y.; Lin, M.-I.; Hsu, H.-T.; Wu, P.-Y.; Wu, C.-J.; Liu, H.-W.; Lin, F.-Y.; Lin, G.; et al. E339-R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 16515–16520.
- Wanitchang, A.; Narkpuk, J.; Jongkaewwattana, A. Nuclear import of influenza B virus nucleoprotein: Involvement of an N-terminal nuclear localization signal and a cleavage-protection motif. Virology 2013, 443, 59–68.
- Sherry, L.; Smith, M.; Davidson, S.; Jackson, D. The N terminus of the influenza B virus nucleoprotein is essential for virus viability, nuclear localization, and optimal transcription and replication of the viral genome. J. Virol. 2014, 88, 12326–12338.
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366.
- Wakai, C.; Iwama, M.; Mizumoto, K.; Nagata, K. Recognition of cap structure by influenza B virus RNA polymerase is less dependent on the methyl residue than recognition by influenza A virus polymerase. J. Virol. 2011, 85, 7504–7512.
- Dauber, B.; Heins, G.; Wolff, T. The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta interferon induction. J. Virol. 2004, 78, 1865–1872.
- Guan, R.; Ma, L.-C.; Leonard, P.G.; Amer, B.R.; Sridharan, H.; Zhao, C.; Krug, R.M.; Montelione, G.T. Structural basis for the sequence-specific recognition of human ISG15 by the NS1 protein of influenza B virus. Proc. Natl. Acad. Sci. USA 2011, 108, 13468–13473.
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395.
- Donelan, N.R.; Dauber, B.; Wang, X.; Basler, C.F.; Wolff, T.; García-Sastre, A. The N-and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 2004, 78, 11574–11582.
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328.
- Paragas, J.; Talon, J.; O’Neill, R.E.; Anderson, D.K.; García-Sastre, A.; Palese, P. Influenza B and C virus NEP (NS2) proteins possess nuclear export activities. J. Virol. 2001, 75, 7375–7383.
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21.
- Crow, M.; Deng, T.; Addley, M.; Brownlee, G.G. Mutational analysis of the influenza virus cRNA promoter and identification of nucleotides critical for replication. J. Virol. 2004, 78, 6263–6270.
- Plotch, S.J.; Tomasz, J.; Krug, R.M. Absence of detectable capping and methylating enzymes in influenza virions. J. Virol. 1978, 28, 75–83.
- Dhar, R.; Chanock, R.M.; Lai, C.J. Nonviral oligonucleotides at the 5′ terminus of cytoplasmic influenza viral mRNA deduced from cloned complete genomic sequences. Cell 1980, 21, 495–500.
- Li, M.L.; Rao, P.; Krug, R.M. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 2001, 20, 2078–2086.
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 2005, 79, 5812–5818.
- Nayak, D.P.; Balogun, R.A.; Yamada, H.; Zhou, Z.H.; Barman, S. Influenza virus morphogenesis and budding. Virus Res. 2009, 143, 147–161.
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153.
This entry is offline, you can click here to edit this entry!