Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
The TAM (TYRO3, MERTK, and AXL) family of receptor tyrosine kinases are pleiotropic regulators of adult tissue homeostasis maintaining organ integrity and self-renewal. Disruption of their homeostatic balance fosters pathological conditions like autoinflammatory or degenerative diseases including rheumatoid arthritis, lupus erythematodes, or liver fibrosis. Moreover, TAM receptors exhibit prominent cell-transforming properties, promoting tumor progression, metastasis, and therapy resistance in various cancer entities. Emerging evidence shows that TAM receptors are involved in bone homeostasis by regulating osteoblastic bone formation and osteoclastic bone resorption. Therefore, TAM receptors emerge as new key players of the regulatory cytokine network of osteoblasts and osteoclasts and represent accessible targets for pharmacologic therapy for a broad set of different bone diseases, including primary and metastatic bone tumors, rheumatoid arthritis, or osteoporosis.
- TAM receptors
- osteoblasts
- osteoclasts
- bone
- TYRO3
- AXL
- MERTK
1. Introduction
TYRO3, AXL, and MERTK are members of the TAM family of receptor tyrosine kinases and are widely expressed throughout the body [1]. They share a similar structure comprised of an extracellular domain containing two immunoglobulin (Ig)-like repeats and two fibronectin type III (FNIII) repeats. The extracellular domain, which allows receptor–ligand interaction, is followed by a transmembrane domain and a cytoplasmic tyrosine kinase domain [2]. The most studied TAM receptor ligands are growth arrest-specific 6 (GAS6) and protein S (PROS1), which share similar structural motifs and are vitamin K-dependent [3]. Both GAS6 and PROS1 comprise three main elements: an N-terminal γ-carboxyglutamic acid (Gla) domain, which regulates Ca2+-dependent binding to phosphatidylserine (PtdSer); four epidermal growth factor (EGF)-like repeats; and a C-terminal sex hormone-binding globulin (SHBG) domain [4,5]. The SHBG domain mediates receptor–ligand binding through its two laminin G-like (LG) domains.
All three TAM receptors are activated by GAS6, which binds receptors with different affinities (AXL > TXRO3 > MERTK), whereas PROS1 activates TYRO3 and MERTK but not AXL [5,6]. TAM receptors are activated by binding of the ligand LG domains to Ig-like domains of the receptor. Optimal receptor–ligand interaction requires the presence of Ca2+ and PtdSer-presenting membrane (i.e., apoptotic cells or enveloped virus) [5]. Activation of the receptor results in receptor dimerization, autophosphorylation of kinase domains, and activation of intracellular signaling cascades [2]. TAM receptors mediate different cellular processes including growth, survival, differentiation, adhesion, migration, and apoptosis [1,2,4].
TAM receptors play a crucial role in physiological tissue homeostasis and inflammation by mediating the clearance of apoptotic cells (such as aged or damaged cells) by professional phagocytes or epithelial cells [7,8,9,10,11,12,13]. Apoptotic cells expose PtdSer on their outer surface, which can bind to TAM receptor ligands [14]. Thereby, GAS6 and PROS1 can function as bridging molecules between phagocytes and apoptotic cells and induce TAM receptor activation resulting in phagocytosis [2]. The process of apoptotic cell engulfment is called efferocytosis, which is a crucial step for the resolution of inflammation [14]. Besides their physiological functions, TAM receptors and their ligands have been associated with chronic inflammatory and autoimmune diseases, such as multiple sclerosis, various rheumatic diseases, and systemic lupus erythematosus [11,15,16,17]. Moreover, TAM receptors are frequently overexpressed in human cancers (such as breast, lung, gastric, metastatic colon, and prostate tumors) and are associated with chemo-resistance, metastasis, and poor prognosis [18,19,20,21,22,23,24].
Recent studies have shown that TAM receptors are involved in bone homeostasis, suggesting that they can be therapeutically targeted in bone disorders [25,26,27]. Bone is a mineralized connective tissue that provides mechanical support for the body and protects internal organs [28]. Besides these functions, it serves as a source of cytokines, hormones, and growth factors, which provides an ideal environment for hematopoietic stem cells (HSCs) [29]. Bone tissue is highly dynamic and continuously undergoes remodeling to maintain bone strength and mineral homeostasis through the coordinated action of bone-forming osteoblasts, bone-resorbing osteoclasts, and osteocytes [30]. Osteoblasts derive from bone marrow-resident mesenchymal stem cells (MSCs), which can differentiate into adipocytes, chondrocytes, osteoblasts, and vascular smooth muscle cells. Osteoblastogenesis is mediated by the induction of master transcription factors runt-related transcription factor 2 (RUNX2) and transcription factor Sp7 (OSTERIX) [30]. Mature osteoblasts secrete an unmineralized bone matrix consisting of type I collagen, which is mineralized by the deposition of calcium and hydroxyapatite [31]. In the termination phase, osteoblasts either undergo apoptosis and become bone lining cells or are embedded into the bone matrix and terminally differentiate into residing osteocytes [32]. Osteocytes are the most abundant and long-lived cells in the bone and orchestrate bone remodeling by sensing and transducing mechanical strains into biochemical signals [33]. Osteoclasts are multinucleated cells and derive from mononuclear HSCs, particularly bone marrow monocyte–macrophage precursors [34]. The remodeling process starts with the recruitment and activation of osteoclast precursor cells [30]. Active osteoclasts attach to the bone and degrade the bone matrix through acidification and the production of enzymes like tartrate-resistant acid phosphatase (TRAP), cathepsin K, or matrix metalloproteinases [32,34]. Following bone resorption, osteoclasts undergo apoptosis and osteoblasts deposit new bone before they become bone lining cells or osteocytes [30]. The fine equilibrium between bone resorption and bone formation is crucial as excessive bone resorption by osteoclasts with insufficient newly formed bone causes osteoporosis. On the contrary, excessive bone formation versus resorption may result in osteopetrosis [32]. The physiological balance in bone remodeling is regulated by different local and systemic factors including biomechanical load, hormones, cytokines, and chemokines [30,32].
2. Role of TAM Receptors in Bone Remodeling
2.1. TAM Receptors in Osteoclasts
Key molecular signals of osteoclastogenesis are mediated by the macrophage colony-stimulating factor (M-CSF) and the receptor activator of NF-κB ligand (RANKL). In early osteoclast differentiation, M-CSF activates mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and phosphoinositide 3-kinases (PI3K)/protein kinase B (AKT) pathways, promoting proliferation, survival, and differentiation. RANKL promotes MAPK/p38/c-Jun N-terminal kinases (JNK), AKT, guanine nucleotide exchange factor VAV3, proto-oncogene tyrosine-protein kinase Src (c-SRC), and tumor necrosis factor receptor-associated factor 6 (TRAF6)/NF-κB signaling cascades to induce microphthalmia-associated transcription factor (MITF), activator protein 1 (AP1), cycling adenosin monophosphate response element binding protein (CREB), and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) activation to promote proliferation, survival, motility, adhesion, and differentiation of osteoclast precursors into mature osteoclasts. RANKL signaling can be further strengthened by costimulatory signaling pathways like triggering receptor expressed on myeloid cells 2 (TREM2) or osteoclast-associated receptor (OSCAR) to induce tyro protein tyrosine kinase-binding protein (DAP12)/Fc receptor common γ subunit (FcRγ)-spleen tyrosine kinase (Syk) phospholipase C-(PLCγ) signaling to activate calcium signaling and NFATc1 induction [35].
The role of AXL in osteoclast biology is largely unknown. It was recently shown that AXL is expressed by osteoclast precursors and contributes to osteoclast differentiation in vitro via upregulation of monocyte chemoattractant protein 1 (MCP-1), a regulator of osteoclast cell–cell fusion. Treatment of osteoclast cultures with the small molecule AXL inhibitor Bemcentinib blocked osteoclast differentiation [36]. In contrast, another study showed that treatment of healthy mice with the pan-TAM receptor inhibitor BMS-777607 had no effect on osteoclast numbers on the bone surface [37]. Transgenic animal models with osteoclast-specific knockout are needed to further explore the role of AXL in osteoclast biology.
The first authors showing an implication of TYRO3 in osteoclast differentiation were Nakamura et al. They observed TYRO3 expression in multinucleated osteoclasts in vivo and measured increased bone-resorbing activity of osteoclasts induced by PROS1 and GAS6, whereas osteoclast differentiation was not affected [38]. Another study confirmed that GAS6 did not affect the differentiation or survival of osteoclasts, but stimulated osteoclast bone resorptive function. Mechanistically, they observed that GAS6 induced TYRO3 phosphorylation and ERK signaling, while inhibition of ERK by PD98059 abrogated the GAS6-mediated effect [39]. However, the role of ERK in osteoclast biology is controversial. Germline deletion of Erk1 (Erk1−/−) and hematopoietic Erk2 conditional knockout (Mx1-Cre+Erk2flox/flox) impaired osteoclast formation and function and increased bone mass by decreasing osteoclast formation in vivo [40]. In contrast, a study showed that ERK inhibition increased osteoclastogenesis in RAW 264.7 osteoclast precursor cell line by stimulating AMP-activated protein kinase (AMPK) activation and RANK expression and inhibiting anti-osteoclastogenic factor expression [41]. Therefore, the pro-osteoclastogenic function of ERK might be restricted to early osteoclast precursors. Ruiz-Heiland et al. showed that GAS6 can indeed promote osteoclast differentiation, but these effects were restricted to the presence of low RANKL concentrations. In the absence of RANKL or with high RANKL concentrations, they did not observe the effects of GAS6 on osteoclast differentiation. Ex vivo osteoclast cultures of bone marrow macrophages from Tyro3−/− mice showed decreased osteoclast differentiation and Tyro3−/− mice exhibited increased bone mass and reduced osteoclast numbers in vivo [42].
Although the literature shows a pro-osteoclastogenic role of TYRO3, its biological function appears complex. The pro-osteoclastogenic action of TYRO3 and especially GAS6 in RANKL-mediated osteoclast differentiation seems to be neglectable in vitro. In line with that, another study showed that treatment of osteoclast cultures with soluble TYRO3 led to increased osteoclast differentiation [43]. As soluble TAM receptors serve as extracellular traps for TAM ligands, these results suggest that PROS1 or GAS6 might dampen osteoclastogenesis.
Another study detected increased TYRO3 expression by FACS analysis on the surface of pro-osteoclastogenic CD14+CD16− monocyte subpopulation in the blood of rheumatoid arthritis patients, indicating a role for TYRO3 in monocytic osteoclast precursor cells in the circulation [44]. During the development of the bone, osteoclasts derive from erythromyeloid progenitors (EMPs) from the blood islands of the yolk sac. In postnatal life, HSC-derived precursors give rise to osteoclasts [45,46]. Although the anatomical site of the main osteoclast precursor cell pool is still under debate, it is acknowledged that osteoclasts derive mainly from a pool of primed monocytic cells in the circulation. The concept of circulatory osteoclast precursors requires highly dynamic and mobile cells. The migratory mechanisms in systemic circulation and homing into bone spaces are critical points of control for osteoclast formation and bone homeostasis. Entry and exit from the bone marrow cavity are regulated by several chemokines and signaling molecules, such as chemokine (C-X-C motif) ligand (CXCL) 12, CC chemokine ligands (CCL)19, CCL21, CX3CL1, and sphingosine-1-phosphate (S1P) [47,48,49,50]. S1P modulates osteoclast precursor cell migration by two counteracting receptors, circulation-attractive S1P receptor 1 (S1PR1) and bone tropic S1PR2, regulating fine-tuned entrance into and exit from the bones [51,52]. Researchers recently showed that the TAM receptor MERTK might be involved in osteoclast precursor cell homing and motility [25,26]. Researchers demonstrated that myeloid cell-specific deletion of MERTK (LysM-cre+Mertkflox/flox) increased bone mass and reduced osteoclastogenesis in vivo [26]. Mechanistically, deletion of MERTK in myeloid cells decreased TRAP+ mononuclear cells in the bone marrow. Furthermore, osteoclast precursor cells lacking MERTK showed decreased motility via the small GTPase Ras homolog family member A (RHOA), suggesting that MERTK regulates osteoclast precursor cell migration and homing to bone marrow cavity [26]. RHOA is a key regulator of osteoclast differentiation and function. It was found to be responsible for C-C chemokine receptor type 7 (CCR7)-dependent migration and chemotaxis, as well as S1PR2-mediated chemorepulsion of monocytic osteoclast precursors [49,51,53]. In mature osteoclasts, RHOA was found to regulate bone resorptive activity [53,54,55]. The precise role of MERTK and TYRO3 in osteoclast differentiation, migration, and function will be the subject of further studies.
Next to that, TAM receptors play a critical role in macrophages, which share a common hematopoietic origin with osteoclasts and are key effector cells in innate and adaptive immunity [56]. TAM receptors are differentially expressed in macrophage development and maturation into their subsequent subpopulations M1 and M2 [57]. M1 and M2 macrophages can be distinguished by their cytokine profiles, which exert opposite effects on osteoclasts. “Classically activated” M1 macrophages secrete pro-inflammatory cytokines that promote osteoclastogenesis, such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), IL-1β, IL-15, and IL-18, while “Alternatively activated” M2 macrophages secrete mostly cytokines, which can inhibit osteoclastic bone resorption such as transforming growth factor β (TGF-β) and IL-10 [58].
However, the influence of M1 vs. M2 macrophage polarization on osteoclastogenesis is under debate. It was shown that RANKL increases M1 macrophages that reside near the growth plate in high proximity to osteoclasts and osteoblasts. Furthermore, treatment with RANKL increased the expression of pro-inflammatory cytokines and inducible nitric oxide synthase (iNOS) in macrophages. The authors suggest that M1 macrophages may finally turn into osteoclasts through induction of RANKL [59]. In contrast, another study showed that in osteoporosis, M2 macrophages but not M1 macrophages differentiate into functional osteoclasts in the presence of RANKL and that estrogen protects M2 macrophages from RANKL stimulation [60]. It has been also shown that M1 macrophages significantly reduced osteoclastogenesis by downregulating master transcription factor NFATc1 and increasing the apoptosis of osteoclasts. This was confirmed in a functional mouse model of ligature-induced periodontitis. M1 and M2 macrophages were adoptively transferred and reduced osteoclastogenesis was observed by the presence of M1 macrophages. This led to the hypothesis that induction of M1 macrophage polarization could be an attractive treatment strategy for reducing osteoclastogenesis in bone diseases [61].
Data on the role of TYRO3 in macrophage polarization are scarce with one study showing that TYRO3 can inhibit lipopolysaccharide (LPS) and interferon-gamma (IFN-γ)-induced M1 polarization of peritoneal and tumor-derived mouse macrophages [62]. The role of AXL is controversial with studies showing that induction of M1 macrophage phenotype, but also M2 macrophage signature, can be induced by AXL depending on the cytokine environment and disease context. Additionally, AXL contributes to efferocytosis in the context of inflammatory environments [63,64,65,66]. In contrast, MERTK is strongly connected to M2 macrophage polarization status and is essential for efferocytosis predominantly in anti-inflammatory environments [67,68,69,70].
If and how TAM receptors influence osteoclastogenesis and bone resorption by macrophage polarization is unknown. However, inhibition of MERTK might be an attractive treatment strategy for targeting macrophages to induce M1 polarization, which may inhibit osteoclastogenesis. Consistently, researchers detected reduced TRAP+ mononuclear cells induced by silencing MERTK genetically, as well as by targeting with a pharmacologic compound. As bone marrow macrophages express TRAP, these effects might have been mediated by altered macrophage polarization, composition, or numbers. Figure 1 summarizes the proposed mechanism of regulation of osteoclast formation by MERTK and TYRO3 (Figure 1a,b).
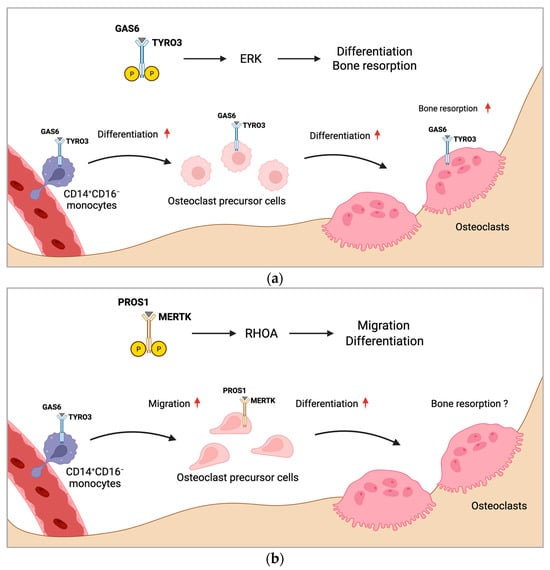
Figure 1. Proposed mechanism of regulation of osteoclast formation by MERTK and TYRO3. (a) CD14+CD16− monocytes with increased TYRO3 expression are the main source of osteoclast precursors. GAS6 induces TYRO3 phosphorylation and ERK signaling, promoting osteoclast differentiation. Activation of TYRO3 increases bone resorptive activity of osteoclasts. (b) MERTK induces RHOA signaling in osteoclast precursor cells leading to enhanced osteoclast differentiation and migration. Rounded arrows indicate differentiation steps in osteoclastogenesis. Straight horizontal arrows indicate TAM receptor signaling pathway leading to different biological functions in osteoclastogenesis. Straight vertical arrows show upregulation of indicated biological functions. The role of MERTK in osteoclastic bone resorption is unknown (?). Created with BioRender.com.
2.2. TAM Receptors in Osteoblasts
The osteogenic fate of MSCs is tightly controlled by factors present in the bone marrow microenvironment. The classical osteogenic differentiation pathways are Wnt/β-catenin and bone morphogenic protein (BMP). Furthermore, numerous hormones impact osteoblast function including insulin-like growth factor type 1 (IGF-1), parathyroid hormone (PTH), PTH-related peptide (PTHrP), active vitamin D (1,25(OH)2D3), leptin, and glucocorticoids. Other important proteins of the osteoblast signaling network are members of the fibroblast growth factor (FGF), sonic hedgehog (SHH), ephrin, and notch family [71].
The first study demonstrating that TAM receptors are potentially involved in osteogenesis showed that AXL modulates the osteogenic differentiation of pericytes [72]. Pericytes are perivascular cells that wrap around blood capillaries providing a source of undifferentiated mesenchymal cells, which can differentiate into other cell types, such as chondrocytes and osteoblasts, and induce ectopic calcification and osteogenesis [73]. The authors showed that AXL is downregulated during osteogenic differentiation. Inhibition of GAS6-AXL signaling enhanced mineralization of pericyte nodules in vitro, but the number of mineralized nodules was decreased [72]. Another study showed that AXL prevents calcified matrix deposition by vascular smooth muscle cells by inducing the PI3K-AKT pathway and inhibiting apoptotic caspase signaling [74]. In contrast, a recent study suggested that inhibition of AXL may reduce the formation of new bone, shown by slightly decreased matrix mineralization in osteoblast cultures upon AXL blockade [27]. The PI3K-AKT pathway has crucial osteoblast-specific effects, promoting proliferation, survival, and differentiation. AKT-1 knockout mice were small with reduced bone mineral density, whereas AKT-1/AKT-2 double knockouts exhibited a strong phenotype with dwarfism and negligible ossification, and died shortly after birth [75,76]. Conditional knockout of phosphatase and tensin homolog (PTEN), which negatively regulates AKT signaling, increased bone volume in mice by promoting osteoblast function and decreasing osteoblast apoptosis [77]. The role of AXL in bone development and homeostasis is largely unknown. Given its ability to increase bone nodule formation, induce the PI3K-AKT pathway, and inhibit caspase signaling, AXL might promote osteoblast differentiation by increasing proliferation and survival. Its inhibitory effect on mineralization suggests a negative regulatory role in mature osteoblast function, which should be explored in further studies.
Researchers recently demonstrated that MERTK and TYRO3 regulate osteoblastic bone remodeling. By using osteoblast-specific knockout mouse models (Col1a1-2,3kb-cre+Mertkflox/flox and Col1a1-2,3kb-cre+Tyro3flox/flox), researchers identified MERTK as a negative regulator of bone mass, whereas TYRO3 enhanced osteoblast differentiation and bone formation. Treatment of healthy mice with the MERTK-specific inhibitor R992 increased bone mass by enhancing osteoblastic bone formation. Mechanistically, researchers found that MERTK induces the RHOA-Rho-associated protein kinase (ROCK)-MYOSIN II pathway, whereas TYRO3 inhibits RHOA-MYOSIN II signaling. Thereby, MERTK induced cytoskeletal remodeling, which promoted cellular contraction and stress fiber formation, whereas TYRO3 stimulated opposite effects [25]. The promising role of MERTK as a target supporting osteoblasts was recently underlined by another study by Decker et al. It was demonstrated that inhibition of MERTK primed alveolar bone mesenchymal stem cells to differentiate into matrix-producing osteoblasts by activating WNT signaling [27].
The WNT/β-CATENIN pathway is the key pathway for osteoblast differentiation. Extracellular ligands of the WNT family glycoproteins (e.g., WNT1, WNT5a, WNT7b, WNT10b, and WNT16) bind to the seven-pass transmembrane G-protein-coupled receptors of the frizzled (FZD) family and its co-receptor of the arrow/low-density lipoprotein receptor-related protein (Lrp) family (e.g., LRP5 and LRP6). The WNT extracellular antagonists Dickkopf (DKK1) and Sclerostin (SOST) represent negative regulators of osteoblast differentiation and bone mass, which are emerging promising therapeutic targets for osteoanabolic therapy. The activation of canonical WNT signaling leads to stabilization and translocation of intracellular β-CATENIN into the nucleus to regulate gene transcription of lymphoid-enhancing factor/T-cell factor (LEF/TCF) to stimulate expression of osteoblast target genes. Cytoplasmic β-CATENIN is regulated by phosphorylation by a protein degradation complex composed of glycogen synthase kinase 3β (GSK3β), axis inhibition protein (AXIN), and adenomatous polyposis coli (APC). GSK3β phosphorylates and tags β-CATENIN for ubiquitination and proteasomal degradation [78].
The RHOA-ROCK pathway has recently emerged as a strong inhibitory pathway for WNT signaling, thereby negatively regulating osteoblast differentiation. It was shown that RHOA knockout in osteoblasts increases bone mass while overexpression decreases it. RHOA-ROCK activation in osteoblasts activated Janus kinase (JAK)1/2, which directly phosphorylated GSK3β, resulting in GSK3β activation and subsequent β-CATENIN destabilization, equalizing osteoblast-promoting wingless-related integration site (WNT) signaling. RHOA loss of function interacted genetically with DKK1 gain of function to rescue the severe limb truncation phenotype in mouse embryonic limb bud ectoderms [79]. Furthermore, another study showed that RHOA-ROCK inhibits IGF-1 signaling in osteoblasts. IGF-1 is one of the most abundant growth factors deposited in the bone matrix. RHOA-ROCK inhibited phosphorylation of insulin receptor substrate 1 (IRS-1) and AKT, key effectors of the IGF-1/IRS-1/PI3K/AKT pathway in osteoblasts [80].
MERTK emerges as a potential key regulator of WNT/β-CATENIN, as well as IGF-1 signaling in osteoblasts via the RHOA-ROCK pathway. As MERTK acts via ROCK downstream of key extracellular osteogenic signals, including members of the WNT family or IGF-1, MERTK could represent a suppressor of osteoblastogenesis independent of important osteoblast growth factors. It was found that the ability of RHOA-ROCK to inhibit WNT signaling is lower in physiological settings in contrast to pathological conditions like age-related bone loss [79]. Therefore, blocking MERTK might be a tool to treat osteopenic bone disorders. In contrast, TYRO3 agonists could prevent RHOA-ROCK signaling in osteoblasts, thereby improving osteoblast function and bone health. Romosozumab is approved for osteoporosis and targets Sclerostin, thereby increasing WNT signaling in osteoblasts [81,82]. As MERTK-RHOA-ROCK signaling interferes with WNT signaling downstream of Sclerostin, activation of MERTK could represent a potential resistance mechanism for Sclerostin-directed therapy in osteoporosis and other bone diseases.
Studies investigating the function of TAM receptor ligands GAS6 and PROS1 in bone homeostasis are scarce. GAS6 and PROS1 are both highly expressed by osteoblasts and PROS1 was found to inhibit osteoblastogenesis via MERTK in vitro [25]. Nevertheless, PROS1 might also promote bone formation via binding to TYRO3 in vivo. The role of GAS6 in osteoblast differentiation and function is unknown. Additional functional studies are needed to widen the knowledge regarding TAM receptor ligands as part of the osteoblast signaling network and how they might contribute to bone diseases. Figure 2 shows the possible interaction of TAM receptors with important members of the osteoblast signaling network (Figure 2).
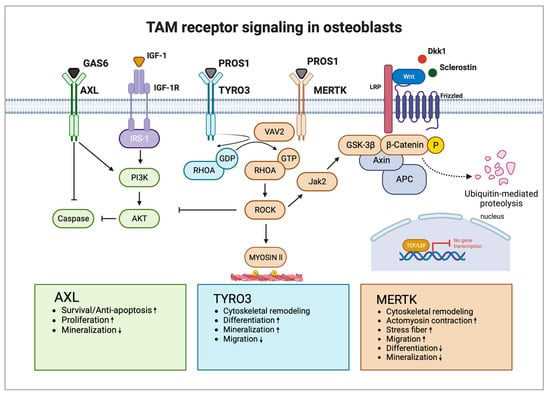
Figure 2. TAM receptors and their downstream signaling pathways in osteoblasts. AXL induces osteoblast proliferation and survival via activation of the PI3K-AKT pathway and inhibition of caspase signaling but inhibits osteoblast mineralization. TYRO3 inhibits RHOA signaling in osteoblasts, promoting cytoskeletal remodeling leading to enhanced differentiation and mineralization but reduced migration. MERTK induces RHOA-ROCK signaling in osteoblasts, which negatively regulates important osteoblastogenesis pathways WNT and IGF-1. Activation of MERTK negatively regulates differentiation and mineralization but promotes migration. Main pathways and biological functions induced by AXL are coloured in green, TYRO3 in turquoise and MERTK in brown. Arrows illustrate activation and blunt arrows silencing of indicated pathways. Boxes at the bottom highlight biological functions of AXL, TYRO3 and MERTK in osteoblasts. Arrow up: Increased function; Arrow down: Decreased function. IGF-1R: IGF-1 receptor; GDP: guanosine diphosphate; and GTP: guanosine triphosphate. Created with BioRender.com.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010233
This entry is offline, you can click here to edit this entry!