Medulloblastoma is the most prevalent intracerebellar pediatric brain tumor, accounting for approximately 20% of all childhood brain tumors and over 60% of embryonal brain tumors [
1]. Medulloblastoma is classified into four major molecularly diverse subgroups including wingless (WNT), Sonic hedgehog (SHH, p53 mutant and p53 wild type), Group 3, and Group 4 medulloblastomas [
2,
3,
4]. The WNT subgroup comprises approximately 10% of the medulloblastoma cases and has the most favorable clinical outcomes, with a 5-year overall survival surpassing 95% [
5,
6,
7]. The SHH subgroup typically displays deregulation of the SHH signaling pathway and represents approximately one-third of childhood patients with medulloblastomas [
2,
8]. Group 3 medulloblastomas often exhibit MYC overexpression and have the most dismal clinical diagnosis of the four medulloblastoma subgroups, with a survival rate of less than 60%. MYC-driven medulloblastomas have extreme metastatic potential and are often resistant to multipronged treatment [
9,
10,
11]. Group 4 is the most prevalent subgroup, accounting for nearly 40% of all medulloblastoma tumors, and is normally seen in children aged 5–10 years and rarely in infants [
2]. Although progress has been made in understanding medulloblastoma at the molecular and genetic level, comparatively few targeted therapies have achieved clinical success. Current therapies for medulloblastoma have progressed in favor of patient survival to about 70% [
8]. However, this comes with consequences, as standard treatment or medications like chemotherapy, brain and spinal cord radiation, and surgical removal leave patients at risk for permanent mental disabilities [
1,
12].
Post-translational modification (PTM) is one targetable regulatory mechanism of MYC and other proteins, with the potential to be developed therapeutically. While the roles of PTMs like phosphorylation [
13], ubiquitinoylation [
14], and acetylation [
15] in controlling these proteins responsible for medulloblastoma have received significant attention, arginine methylation has only recently been investigated. Arginine methylation is one of the common PTM processes that are catalyzed by a member of the protein arginine methyltransferase (PRMT) family; this group of nine enzymes is responsible for the methylation of arginine, using S-adenosylmethionine (SAM) as a methyl group donor. The physiological control of many cellular processes, including splicing transcription and mitosis, depends on the activity of PRMT family enzymes [
16]. PRMTs have also been revealed to be involved in the progression of various types of cancers [
17,
18]. In humans, PRMT members can be divided into various classes based on their enzymatic role, i.e., type I (PRMT1-4, PRMT6, and PRMT8) that catalyze the formation of monomethyl arginine (MMA) and asymmetric dimethyl arginine (ADMA); type II (PRMT5 and PRMT9) that catalyze the formation of MMA and symmetric dimethyl arginine (SDMA); and type III (PRMT7) which is responsible for the formation of MMA [
19]. As the most prevalent type II SDMA methyltransferase, PRMT5 forms a heterotetrametric complex with a protein called methylosome protein 50 (MEP50) that can catalyze symmetric demethylation of various histone and non-histone proteins [
20]. Remarkably, PRMT5 was proven to regulate the function of glioma-associated oncogene homolog 1(GLI1) protein in an SHH-responsive cell line [
21]. PRMT5 also represents a requisite driver of tumor progression in SHH-medulloblastoma and MYC-amplified medulloblastoma [
22,
23]. During conversion to malignancy or metastasis, PRMT5 acts as an oncogene. PRMT5 enzyme inhibition or its catalytic depletion frequently reduces or halts cellular proliferation, while its hyperexpression leads to hyper-proliferation [
24,
25,
26]. Consequently, PRMT5 is emerging as a novel target for the treatment of various cancers, including medulloblastoma.
2. PRMT5 Structure, Function, and Localization
2.1. Structure
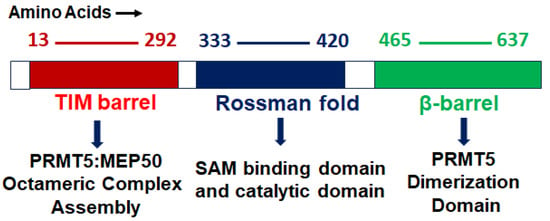
PRMT5 is a primary type II arginine methyl transferase that forms a prominent methylosome complex with distinctive binding oligopeptides, such as the WD (Trp-Asp) repeat-containing 50-kilodalton methylosome protein (MEP50). PRMT5 requires the existence of diverse substrate adapters such as rio-domain-containing protein 1 (RioK1), chloride channel nucleotide-sensitive 1A protein (pIC1n) and cooperator of PRTM5 (COPR5) to detect and catalyze the SDMA on histone and non-histone proteins via PTMs [
31,
32,
33]. PRMT5′s structure consists of a triphosphate isomerase (TIM) barrel, an intermediate Rossmann-fold, and a C-terminal β-barrel [
34]. Four PRMT5 units generate a hetero octameric complex by binding with four MEP50s (
Figure 1). Studies have demonstrated that PRMT5 alone has minimal methyltransferase activity; it must be complexed with MEP50 to achieve normal catalysis of SDMA on proteins [
35]. This could be because MEP50 enhances the stability of PRMT5 for a long time by binding with proteins and acting as a metastable cofactor.
Figure 1. PRMT5 protein structure: structural and functional domains.
2.2. Function
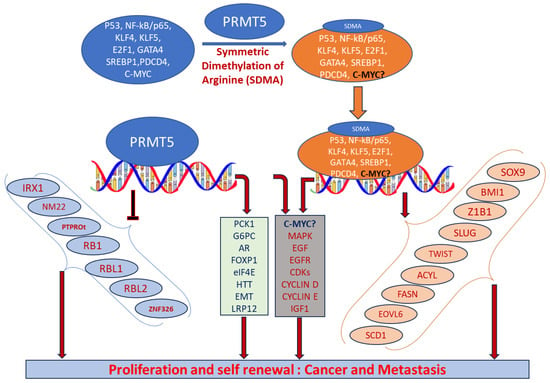
PRMT5 plays a key role in cell functions and processes by regulating the methylation of cellular proteins, which affects oncogenic cellular processes such as cell proliferation and differentiation [
29,
30,
36]. PRMT5 regulates these processes by modifying gene expression to stabilize histones H4R3, H3R2, H3R8, and H2AR3 and non-histone proteins via the SDMA process [
37,
38]. An extensive range of nonhistone proteins have also been revealed as PRMT5 substrates, including androgen receptor (AR), EGFR, GATA4, C-MYC, N-MYC, IL-2, E2F1, GM130, HOXA9, KLF4, KLF5, NOTCH, NFkB(p65), PDCD4, POLR2A, P53, RAF proteins, SPT5, SREBP1a, Sm proteins, nucleolin, and others [
12,
36,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52]. In addition, some substrates such as certain nonhistone oncogenic transcription factors are symmetrically dimethylated by PRMT5. PRMT5-regulated cellular processes are shown in
Figure 2. The importance of arginine methylation by PRMT5 in cancer progression has only recently become apparent [
17]. PRMT5 knockout mice exhibited embryonic lethality, demonstrating the role of PRTM5 in embryonic development and crucial biological functions. In mouse embryonic stem cells (ESCs), PRMT5 maintains pluripotency, whereas in human ESCs, it influences only proliferation [
53,
54]. PRMT5 is needed for neural stem cell persistence and its deletion causes premature death of the mouse by disrupting the development of the central nervous system [
55]. PRMT5 promotes SWI/SNF-mediated chromatin remodeling and controls the process of myogenesis. Deletion of PRMT5 causes an obstacle in developmental processes, uncontrolled proliferation, and impairment of adult tissue differentiation [
53,
54,
55,
56,
57,
58]. Notably, PRMT5 is overexpressed in a number of cancers, including melanoma, multiple myeloma, lymphoma, glioblastoma, breast, lung, pancreas, prostate, ovarian, and colorectal cancers, and high expression of PRMT5 often correlates with poor patient clinical outcomes [
38,
59]. Organ-specific functions of PRMT5 are shown in
Table 1. The higher expression of PRMT5 in cancer is thought to epigenetically suppress tumor suppressor and cell cycle genes [
17,
60]. Recently, the association of PRMT5 with MYC was found in numerous cancers, including brain tumors such as glioblastoma; this association creates abnormalities in MYC function [
61,
62,
63]. Consequently, PRMT5 has been recognized as an oncogenic function and has received extensive interest as a potential target for better clinical outcomes. To this end, numerous potent therapeutic agents have been developed to inhibit PRMT5 and their antitumor effects are now being assessed in preclinical models and clinical trials [
27,
64].
Figure 2. Biological functions of PRMT5 that regulate cellular processes. Elevated expression of PRMT5 can cause post-translational modification of several transcription factors by symmetrically dimethylating arginine residues of proteins and regulate the expression of their corresponding targeted genes. When recruited to the promoter regions of precise target genes in the nucleus, they can promote cell proliferation and tumorigenesis.
2.3. Localization
Cytosolic and nuclear localization of PRMT5 helps to determine its role in the cell. PRMT5 is predominantly localized in the cytoplasm in lung [
65], prostate [
66], and melanoma cancer [
67]. Diffused cellular localization of PRMT5 was confirmed in both the cytoplasm and the nucleus of brain tumor glioblastoma cells [
61]. Cytoplasmic and nuclear localization of PRMT5 has also been confirmed in various preclinical mouse models and primary human cancer tissues [
68]. In adult mice, PRMT5 is expressed predominantly in the nucleus of the neurons in the cerebrum and spinal cord [
55]. Han et al. demonstrated the high expression of PRMT5 as a marker of malignant progression in glioblastoma and its crucial role in tumor growth [
63]. Our lab recently analyzed the localization of the PRMT5 in tumor tissues of medulloblastoma patients as well as in MYC-amplified cell lines. PRMT5 demonstrated predominantly nuclear localization in both HD-MB03 and primary tumor cells [
22].
Table 1. Organ-specific roles of PRMT5.
Abbreviations: AKT-ERK, alpha serine/threonine-protein-extracellular-regulated kinase; AR, androgen receptor; E2F1, E2 promoter binding factor 1; EMT, epithelial–mesenchymal transition; ERG, ETS-related gene; FGFR3, fibroblast growth factor 3; FOXP1, forkhead box protein P1; FUS, fused in sarcoma; GSK3β-NF-kβ, glycogen synthase kinase; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; HTT, huntingtin protein; KLF4, Kruppel-like factor 4; LRP12, low-density lipoprotein receptor-related protein 12; LINC01138, long non-coding RNA; miR-99, microRNA99; mTOR, mammalian target of rapamycin; OCT4, octamer binding protein 4; PKB, protein kinase B; PDCD4, program cell death protein 4; SREBP, sterol regulatory element-binding protein; ZNF326, zinc finger protein 326.
3. PRMT5 Association with MYC-Driven Medulloblastoma
Epigenetic deregulation plays a key role in medulloblastoma tumorigenesis, especially in aggressive Group 3 and Group 4 medulloblastomas [
99,
100,
101,
102], where germline mutations in known cancer predisposition genes are rare. Indeed, epigenetic deregulator or chromatin modifiers, including histone acetylase or methylation/methyltransferase activities, are very common in Group 3 and 4 medulloblastomas compared to other subgroups. This emphasizes the need to discover and understand the pertinent mechanisms of epigenetic regulation or PTMs and the corresponding therapeutic targets. We recently reported that PRTM5 is a critical regulator MYC oncoprotein in an MYC-amplified (Group 3) medulloblastoma [
22]. We found that high levels of PRMT5 not only mirror MYC expression but also correlate with poor outcomes in Group 3 medulloblastoma patients. Mechanistically, we showed that PRMT5 stabilizes the MYC protein by physically interacting with it, raising the intriguing possibility that PRMT5 can regulate MYC function at both the transcriptional and translational/post-translational levels. The exact MYC oncogenic programs regulated by PRMT5 in medulloblastoma are largely unknown. Therefore, exploring the regulation of MYC-driven oncogenic progresses by PRMT5 is crucial to identify effective therapeutics for these high-risk patients.
The involvement of PRMT5 has been verified in the epigenetic regulation of chromatin complexes following interaction with numerous proteins, including transcription factors [
42], and their activities are dysregulated in various cancers [
59]. In recent studies, high levels of PRMT5 and MYC corelate with glioma malignancy [
61,
62,
63]. Furthermore, PRMT5 is physically associated with N-MYC (an MYC homologue) and enhances the stability of N-MYC in neuroblastoma cells [
51]. Nonetheless, the function of PRMT5 and its interaction with MYC in MYC-driven medulloblastoma have not been fully investigated. Favia et al. reported that the association of PRMT1 and PRMT5 with MYC in glioblastoma stem cells resulted in MYC being dimethylated symmetrically and asymmetrically by both enzymes, respectively [
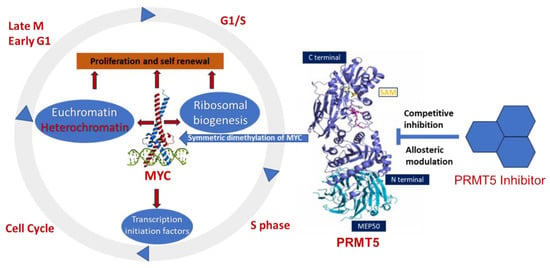
103]. MYC-driven cellular processes resulting from symmetric dimethylation by PRMT5 are shown in
Figure 3. The colocalization of PRMT5 and MYC suggests that PRMT5 forms a complex with MYC and supports its stabilization in MYC-amplified medulloblastoma cells. This physical interaction of PRMT5 and MYC implies a potential role of PRMT5 in medulloblastoma tumorigenesis.
Figure 3. Overexpression of PRMT5 causes symmetric demethylation and stabilization of MYC, leading to reduced apoptosis and enhanced cell proliferation. As indicated, various steps in this process can be modulated by PRMT5 inhibitors.
Highly expressed PRMT5 stabilizes MYC and promotes its expression in medulloblastomas. Studies support the predictive value of PRMT5 overexpression as a biomarker for aggressive tumorigenesis in cancer patients. Knockdown of PRMT5 in medulloblastoma cells suppresses cell growth by diminishing MYC stability, supporting the functional role of the PRMT5–MYC interaction complex in medulloblastoma [
22]. Since MYC and PRMT5 co-expression and colocalization were observed in the nucleus, PRMT5 could also regulate MYC function at the transcriptional level. Further studies are needed to investigate PRMT5’s roles in the regulation of the transcription and translation of MYC.
PRMT5 is a stemness factor crucial in maintaining the balance between quiescence, proliferation, and generation for cancer stem cells and non-cancer cells. The role of PRMT5 in stemness has been demonstrated in embryonic (ESCs) and neural stem cells (NSCs) [
53,
72,
104]. Provided that NCCs or cancer stem cells have a great influence on medulloblastoma recurrence and tumorigenesis, there might be a role for PRMT5 in regulating the self-renewal of tumor initiation in medulloblastoma. Recently, the methylation of stemness factor KLF-4 (Kruppel-like factor-4) by PRTM5 was shown in breast cancer [
96]. This methylation leads to KLF4 protein stabilization, promoting tumorigenesis. In another study, researchers synthesized a novel compound that has the potency to inhibit PRMT5, disrupt the interaction of PRMT5 and KLF4, and suppress breast cancer development [
105]. KLFs are evolutionarily conserved zinc-finger-associated transcription factors with distinct regulatory functions in cell growth, proliferation, and differentiation. Moreover, PRMT5 interacts with KLF5 (another member of KLF family proteins) and accelerates its dimethylation, a mechanism that depends on methyltransferase activity [
81]. Further investigation to understand the mechanism of PRTM5–KLF4/KL5 interactions could uncover another new strategy to elucidate therapeutic targets for MYC-amplified medulloblastoma.
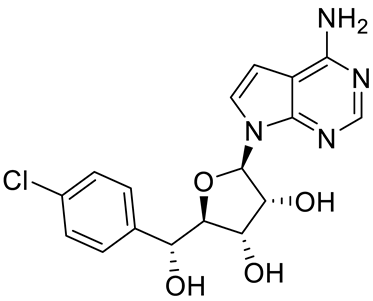
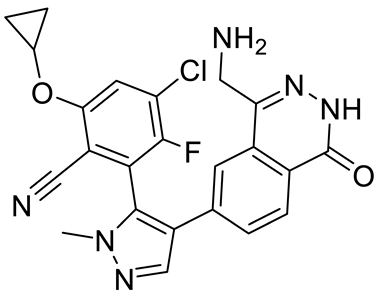
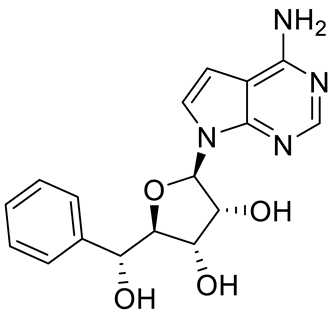
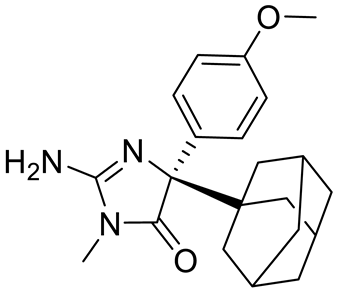
4. Potential Inhibitors of PRMT5
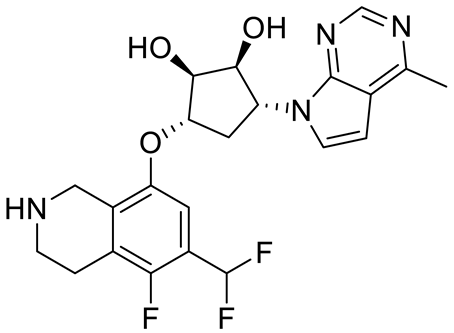
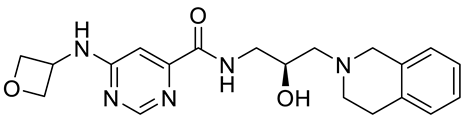
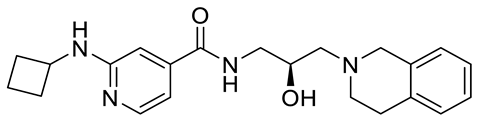
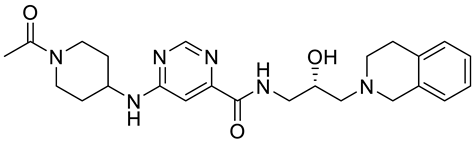
PRMT5 inhibitors have been proven to prevent the growth of cancerous cells in vitro and in vivo. Many PRMT5 inhibitors have entered clinical trials for the treatment of multiple types of cancer [
34,
106,
107]. The pharmacological effects of these inhibitors with their targets in various cancers are summarized in
Table 2, and details about corresponding clinical trials are given in
Table 3.
Table 2. Pharmacologically active PRMT5 inhibitors.
Abbreviations: AML, acute myeloid leukemia; MCL, mantle cell lymphoma; MM, myelomonocytic leukemia; GBM, glioblastoma; TNBC, triple-negative breast cancer, PDX, patient-derived xenograft; DLBCL, diffuse large B cell lymphoma; CNS, central nervous system; MPN, myeloproliferative neoplasm; nM, nano molar; NA, not available; SAM, S-adenosylmethionine; MTA, methylthioadenosine.
Table 3. PRMT5 inhibitors in clinical trials.
|
ClinicalTrials.gov Identifier
|
Name of
Inhibitor
|
Status
|
Disease
|
|
NCT03573310
|
JNJ64619178
|
Phase I
|
Neoplasm solid tumors, non-Hodgkin lymphoma, and myelodysplastic syndrome
|
|
NCT03854227
|
PF06939999
|
Phase I
|
Advance and metastatic solid tumors
|
|
NCT03614728
|
GSK3326595
|
Phase I and II
|
Metastatic solid tumors and acute myeloid leukemia
|
|
NCT02783300
|
GSK3326595
|
Phase I
|
Solid tumors and non-Hodgkin lymphoma
|
|
NCT04676516
|
GSK3326595
|
Phase II
|
Early-stage breast cancer
|
|
NCT03886831
|
PRT543
|
Phase I
|
Advanced solid tumors and hematological malignancies
|
|
NCT05275478
|
TNG908
|
Phase I and II (recruiting)
|
Locally advanced solid tumors
|
|
NCT04089449
|
PRT811
|
Phase I (recruiting)
|
Advanced solid tumors, recurrent glioma, and CNS lymphoma
|
|
NCT05245500
|
MRTX1719
|
Phase I and II (recruiting)
|
Mesothelioma, NSCLC, malignant peripheral nerve sheath tumors, solid tumors, and pancreatic adenocarcinoma
|
|
NCT05094336
|
AMG 193
|
Phase I and II (recruiting)
|
Advanced MTAP-null solid tumors
|
|
NCT05528055
|
SCR6920
|
Phase I (recruiting)
|
Advanced malignant tumors
|
|
NCT05015309
|
SH3765
|
Phase I (not yet Recruiting)
|
Advanced malignant tumors
|