Restrictive cardiomyopathy (RCM) is characterized by restrictive ventricular pathophysiology determined by increased myocardial stiffness. While suspicion of RCM is initially raised by clinical evaluation and supported by electrocardiographic and echocardiographic findings, invasive hemodynamic evaluation is often required for diagnosis and management of patients during follow-up. RCM is commonly associated with a poor prognosis and a high incidence of heart failure, and PH is reported in paediatric patients with RCM. Only a few therapies are available for specific RCM aetiologies. Early referral to centres for advanced heart failure treatment is often necessary.
- restrictive cardiomyopathy
- paediatric
1. Introduction
2. How Common Is RCM?
3. Is Family History and Genetic Testing Important?
4. What Are the Main Clinical Signs and Symptoms of Paediatric RCM?
5. When Should I Suspect Restrictive Phenotype on Electrocardiogram or Imaging Techniques?

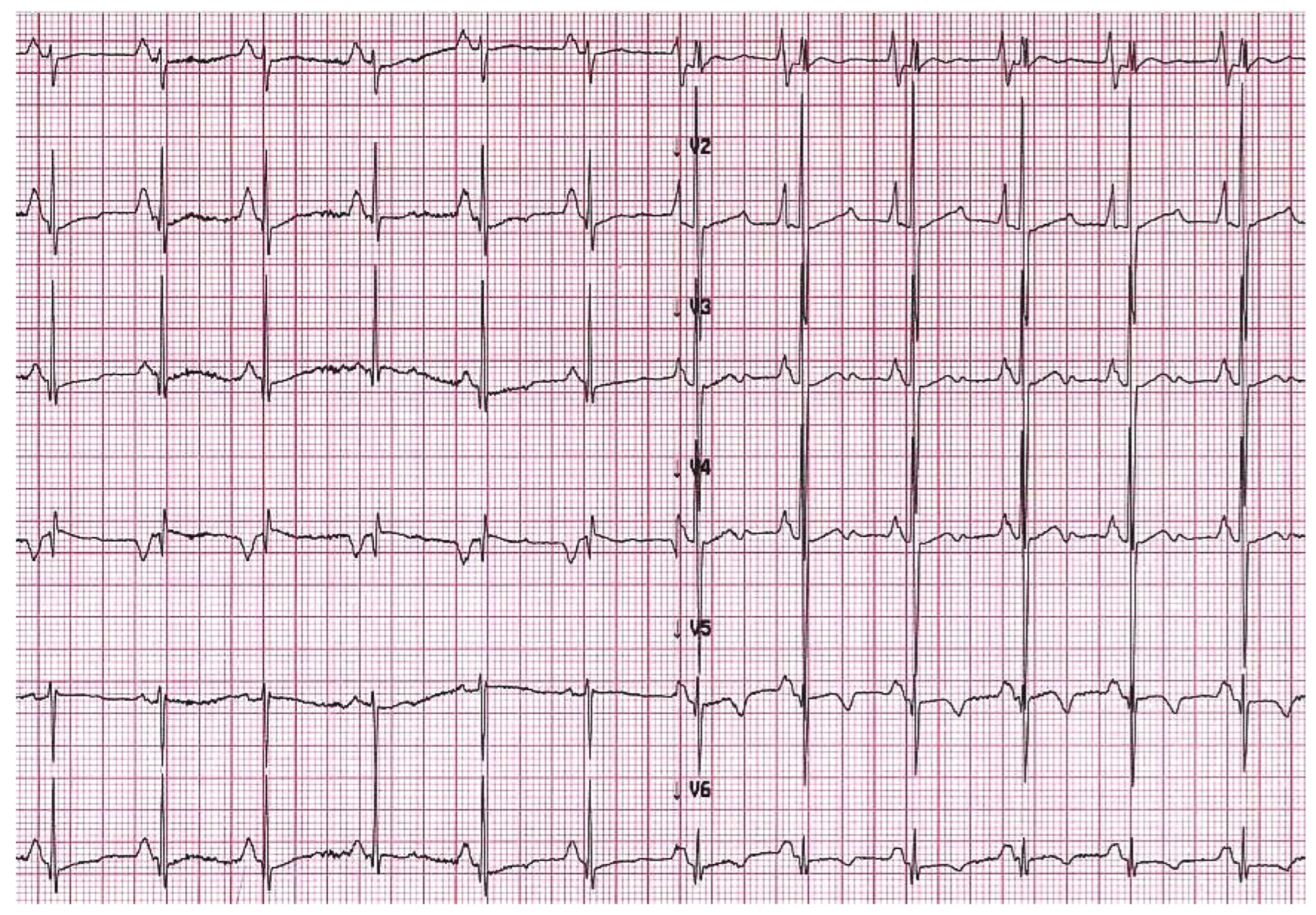

6. What Are the Limitations of ECG and Imaging Techniques?
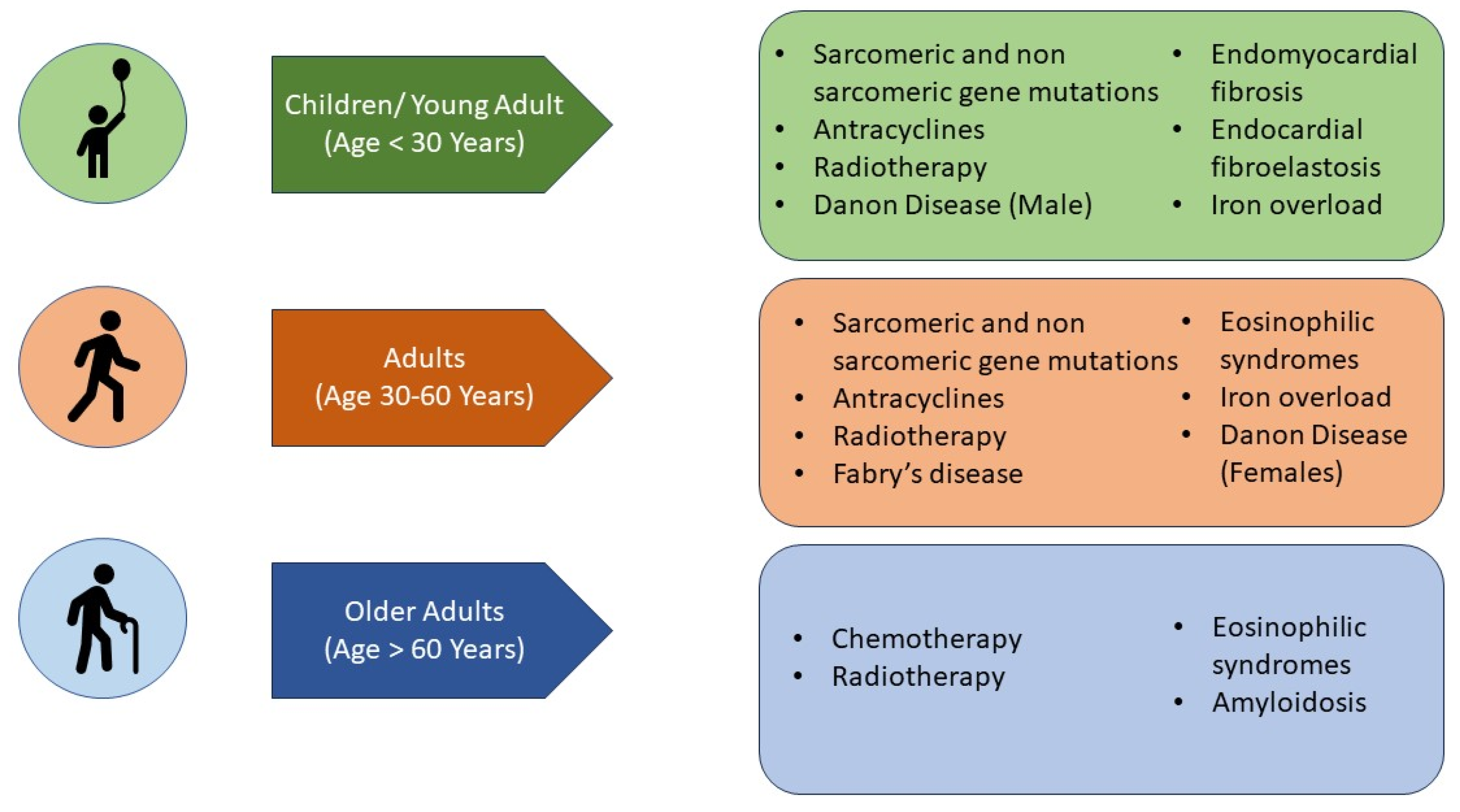
7. What Are the Aetiologies That Mainly Affect the Paediatric Population (Figure 4)?
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics13243666
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; De Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626.
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive cardiomyopathy: Definition and diagnosis. Eur. Heart J. 2022, 43, 4679–4693.
- Denfield, S.W. Overview of pediatric restrictive cardiomyopathy—2021. Prog. Pediatr. Cardiol. 2021, 62, 101415.
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873.
- Ditaranto, R.; Caponetti, A.G.; Ferrara, V.; Parisi, V.; Minnucci, M.; Chiti, C.; Baldassarre, R.; Di Nicola, F.; Bonetti, S.; Hasan, T.; et al. Pediatric Restrictive Cardiomyopathies. Front. Pediatr. 2021, 9, 745365.
- Mocumbi, A.O.; Ferreira, M.B.; Sidi, D.; Yacoub, M.H. A population study of endomyocardial fibrosis in a rural area of Mozambique. N. Engl. J. Med. 2008, 359, 43–49.
- Bukhman, G.; Ziegler, J.; Parry, E. Endomyocardial Fibrosis: Still a Mystery after 60 Years. PLoS Negl. Trop. Dis. 2008, 2, e97.
- Webber, S.A.; Lipshultz, S.E.; Sleeper, L.A.; Lu, M.; Wilkinson, J.D.; Addonizio, L.J.; Canter, C.E.; Colan, S.D.; Everitt, M.D.; Jefferies, J.L.; et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: A report from the Pediatric Cardiomyopathy Registry. Circulation 2012, 126, 1237–1244.
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 819–837.
- Masarone, D.; Valente, F.; Rubino, M.; Vastarella, R.; Gravino, R.; Rea, A.; Russo, M.G.; Pacileo, G.; Limongelli, G. Pediatric Heart Failure: A Practical Guide to Diagnosis and Management. Pediatr. Neonatol. 2017, 58, 303–312.
- Lipshultz, S.E.; Cochran, T.R.; Briston, D.A.; Brown, S.R.; Sambatakos, P.J.; Miller, T.L.; Carrillo, A.A.; Corcia, L.; Sanchez, J.E.; Diamond, M.B.; et al. Pediatric cardiomyopathies: Causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol. 2013, 9, 817–848.
- Quennelle, S.; Bonnet, D. Pediatric heart failure with preserved ejection fraction, a review. Front. Pediatr. 2023, 11, 1137853.
- Sasaki, N.; Garcia, M.; Ko, H.H.; Sharma, S.; Parness, I.A.; Srivastava, S. Applicability of published guidelines for assessment of left ventricular diastolic function in adults to children with restrictive cardiomyopathy: An observational study. Pediatr. Cardiol. 2015, 36, 386–392.
- Barretto, A.C.; Mady, C.; Nussbacher, A.; Ianni, B.M.; Oliveira, S.A.; Jatene, A.; Ramires, J.A. Atrial fibrillation in endomyocardial fibrosis is a marker of worse prognosis. Int. J. Cardiol. 1998, 67, 19–25.
- Muraji, S.; Sumitomo, N.; Imamura, T.; Yasuda, K.; Nishihara, E.; Iwamoto, M.; Tateno, S.; Doi, S.; Hata, T.; Kogaki, S.; et al. Diagnostic value of P-waves in children with idiopathic restrictive cardiomyopathy. Heart Vessel. 2021, 36, 1141–1150.
- Ryan, T.D.; Madueme, P.C.; Jefferies, J.L.; Michelfelder, E.C.; Towbin, J.A.; Woo, J.G.; Sahay, R.D.; King, E.C.; Brown, R.; Moore, R.A.; et al. Utility of Echocardiography in the Assessment of Left Ventricular Diastolic Function and Restrictive Physiology in Children and Young Adults with Restrictive Cardiomyopathy: A Comparative Echocardiography-Catheterization Study. Pediatr. Cardiol. 2017, 38, 381–389.
- Denfield, S.W.; Webber, S.A. Restrictive Cardiomyopathy in Childhood. Heart Fail. Clin. 2010, 6, 445–452.
- Rosales, X.Q.; Moser, S.J.; Tran, T.; McCarthy, B.; Dunn, N.; Habib, P.; Simonetti, O.P.; Mendell, J.R.; Raman, S.V. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J. Cardiovasc. Magn. Reson. 2011, 13, 39.
- de Ville de Goyet, M.; Brichard, B.; Robert, A.; Renard, L.; Veyckemans, F.; Vanhoutte, L.; Moniotte, S. Prospective cardiac MRI for the analysis of biventricular function in children undergoing cancer treatments. Pediatr. Blood Cancer 2015, 62, 867–874.
- Axelsson Raja, A.; Farhad, H.; Valente, A.M.; Couce, J.-P.; Jefferies, J.L.; Bundgaard, H.; Zahka, K.; Lever, H.; Murphy, A.M.; Ashley, E.; et al. Prevalence and Progression of Late Gadolinium Enhancement in Children and Adolescents With Hypertrophic Cardiomyopathy. Circulation 2018, 138, 782–792.
- Garcia, M.J. Constrictive Pericarditis Versus Restrictive Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 67, 2061–2076.
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68.
- Habib, G.; Bucciarelli-Ducci, C.; Caforio, A.L.P.; Cardim, N.; Charron, P.; Cosyns, B.; Dehaene, A.; Derumeaux, G.; Donal, E.; Dweck, M.R.; et al. Multimodality Imaging in Restrictive Cardiomyopathies: An EACVI expert consensus document In collaboration with the “Working Group on myocardial and pericardial diseases” of the European Society of Cardiology Endorsed by The Indian Academy of Echocardiography. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1090–1121.
- Rutakingirwa, M.; Ziegler, J.L.; Newton, R.; Freers, J. Poverty and eosinophilia are risk factors for endomyocardial fibrosis (EMF) in Uganda. Trop. Med. Int. Health 1999, 4, 229–235.
- de Carvalho, F.P.; Azevedo, C.F. Comprehensive Assessment of Endomyocardial Fibrosis with Cardiac MRI: Morphology, Function, and Tissue Characterization. Radiographics 2020, 40, 336–353.
- Ozdemir, D.; Cortopassi, I.O.; McNamara, R.L. An illustrative case of endocardial fibroelastosis and recalcitrant intracardiac thrombosis: A case report. Thromb. J. 2019, 17, 8.
- Harris, L.C.; Nghiem, Q.X. Cardiomyopathies in infants and children. Prog. Cardiovasc. Dis. 1972, 15, 255–287.
- Weixler, V.; Hammer, P.E.; Marx, G.R.; Emani, S.M.; Del Nido, P.J.; Friehs, I. Flow disturbances and progression of endocardial fibroelastosis—A case report. Cardiovasc. Pathol. 2019, 42, 1–3.
- Schryer, M.J.; Karnauchow, P.N. Endocardial fibroelastosis; etiologic and pathogenetic considerations in children. Am. Heart J. 1974, 88, 557–565.
- Lane, K.L.; Herzberg, A.J.; Reimer, K.A.; Bradford, W.D.; Schall, S.A. Endocardial fibroelastosis with coronary artery thromboembolus and myocardial infarction. Clin. Pediatr. 1991, 30, 593–598.