Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Sarcopenia is an age-related condition that involves a progressive decline in muscle mass and function, leading to increased risk of falls, frailty, and mortality. Disruption of the circadian system with age may initiate these pathways in skeletal muscle, preceding the onset of sarcopenia. There is no pharmacological treatment for sarcopenia, only resistance exercise and proper nutrition may delay its onset. Melatonin, derived from tryptophan, emerges as an exceptional candidate for treating sarcopenia due to its chronobiotic, antioxidant, and anti-inflammatory properties.
- aging
- mitochondria
- skeletal muscle
- sarcopenia
- melatonin
- pineal
- clock gene
1. Definition and Primary Characteristics of Sarcopenia
Sarcopenia is a term derived from the Greek σάρξ (sarx, meaning “flesh”), and πενία (penia, meaning “poverty” or “scarcity”). It was first described in the 1980s as an age-related decrease in body muscle mass that affects mobility, nutritional status, and independence [1]. The definition has evolved since then, marked by two recent milestones: the inclusion of muscle function (defined by muscle strength, power, and physical performance) in the concept, as it has been shown to be a clinically more powerful predictor than muscle mass alone; and the recognition of sarcopenia as an independent condition with a code in the International Classification of Diseases-10 (ICD-10) [2][3]. However, most clinicians are still unaware of the condition and the necessary diagnostic tools to identify it.
Even today, the most cited definition for sarcopenia was proposed by the European Working Group on Sarcopenia in Older People (EWGSOP), endorsed by the Asian Working Group on Sarcopenia (AWGS), and updated as EWGSOP2 in January 2019. In this definition, sarcopenia is described as a progressive and widespread disorder of skeletal muscle involving accelerated loss of muscle mass and function, associated with an increase in adverse outcomes such as falls, functional decline, frailty, and mortality [4].
Sarcopenia is a universal phenomenon with a complex and multifactorial etiology. It commonly occurs as an age-related process in older individuals, although it can also occur in middle age in association with a variety of conditions. Factors involved in sarcopenia include aging as a primary cause, but also genetic factors, inactivity or a sedentary lifestyle, nutritional causes (low protein and energy intake, vitamin D deficiency, …), bone and joint diseases, cardiorespiratory disorders, hormonal changes (decrease in testosterone and growth hormone levels), diabetes, neurological diseases, cancer, liver and kidney disorders, and iatrogenic factors (hospitalization, drug-related side effects) [2][5].
In clinical practice, it is established that an individual with low muscle strength and low muscle mass or quality should be diagnosed with sarcopenia. The condition can be better understood as a failure or insufficiency of skeletal muscle [6]. Thus, sarcopenia can manifest acutely (usually in the context of an acute illness or sudden immobility, such as during a hospitalization) or have a more prolonged (chronic) course [2]. Muscle mass and strength (along with bone mineral density) peak in young adulthood and, after a period of stability, begin to gradually decline with age, with a faster decline in strength [7]. However, most cases of sarcopenia go undiagnosed, only coming to attention when relevant symptoms are reported. These symptoms could include falls, weakness, slowness, self-reported loss of muscle mass, or difficulties in performing daily activities. Detecting cases is particularly relevant in care settings where a higher prevalence of sarcopenia might be expected, such as temporary hospital admissions, rehabilitation environments, or nursing homes [8].
Indeed, the detection of sarcopenia is not so straightforward. At times, sarcopenia may be associated with thinness, and it may not be recognized that it can also be present in obesity. Sarcopenic obesity is often identified when a person exhibits both low muscle mass and increased adiposity, but it might go unnoticed when the focus of attention is on obesity, leading to adverse outcomes [9]. Sarcopenia can also be mistaken for other conditions like malnutrition or cachexia, although it is true that it can coexist with both. These conditions involve a reduction in weight and muscle mass, but only sarcopenia entails a decrease in muscle strength with impaired muscle function. Sarcopenia is also closely linked to physical frailty, described as the biological substrate of the frailty phenotype, which involves involuntary weight loss, self-reported exhaustion, weakness (low grip strength), slow walking speed, and low physical activity [10][11]. As we can see, sarcopenia can occur in association with a variety of chronic conditions in middle age, with most patients presenting more than one associated condition.
Research in this area has been challenging for several reasons, including differing opinions on the definition of sarcopenia, the increasing recognition of acute and chronic forms of sarcopenia, the existence of many interactive pathways involved in its pathophysiology, and the impact of related conditions (including those that might mimic sarcopenic symptoms and other existing patient conditions that affect sarcopenia).
2. Synthesis, Metabolism, and Targets of Melatonin
Melatonin, chemically known as N-acetyl-5-methoxytryptamine, is an indoleamine derived from tryptophan. It was first isolated from bovine pineal glands in 1958 by Aaron Lerner, who identified its chemical structure [12]. It is a highly conserved molecule throughout evolution, found in nearly all organisms, from bacteria and plants to invertebrates and mammals [13].
Initially, melatonin was thought to be exclusively produced by the pineal gland and was primarily investigated for its role in regulating circadian rhythms. But melatonin is also synthesized in most tissues and organs of the body, including skeletal muscle, through the same enzymatic processes as in the pineal gland. Therefore, it is differentiated between two types of melatonin: pineal and extrapineal, each with distinct properties, with the latter displaying concentrations typically one or two orders of magnitude higher than pineal melatonin, depending on the tissue. This leads to significantly elevated percentage levels [14][15]. In vertebrate animals, pineal melatonin synthesis occurs during the night, in synchrony with the light–dark cycle and is controlled by the central biological clock in the SCN, as explained earlier. At the cellular level, melatonin synthesis primarily takes place in the mitochondria. It begins with the absorption of tryptophan by pinealocytes from the bloodstream, which is hydroxylated by the enzyme tryptophan hydroxylase (TPH) at position 5, giving rise to 5-hydroxytryptophan. Subsequently, 5-hydroxytryptophan is decarboxylated to form serotonin (also known as 5-hydroxytryptamine) through the action of the enzyme L-amino acid aromatic decarboxylase (AADC). Serotonin is acetylated by the enzyme AANAT, resulting in N-acetylserotonin. Then, N-acetylserotonin is methylated by ASMT, ultimately producing the melatonin molecule [16] (Figure 1). The enzymes AANAT and ASMT are crucial in melatonin synthesis, although there is controversy about which of the two acts as the limiting enzyme in the process [17]. It is important to note that pineal melatonin is not stored within the gland but is released directly into the bloodstream, from where it is distributed throughout the body. In contrast, although the same enzymes are involved in melatonin synthesis in extrapineal tissues, its production does not follow a circadian rhythm. Instead, each tissue produces the necessary amount at any given time, and melatonin remains inside the cell [14][18].
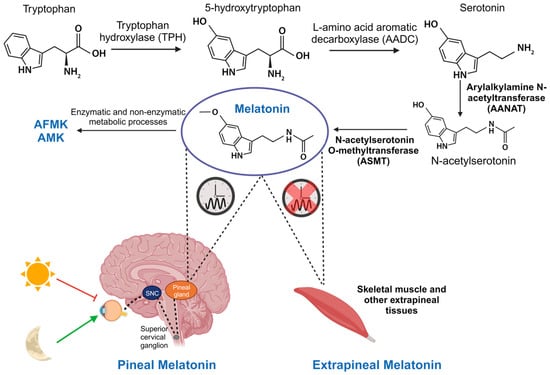
Figure 3. Synthesis of pineal and extrapineal melatonin. In the pineal gland, production of this molecule is nocturnal and regulated by the central biological clock in the SCN. It starts with tryptophan hydrolysis by the enzyme TPH, yielding 5-hydroxytryptophan. AADC then decarboxylates it, creating serotonin. AANAT acetylates serotonin, producing N-acetylserotonin, which is subsequently converted into melatonin by ASMT. Although the same enzymes are involved in its synthesis, in skeletal muscle and other extrapineal tissues, its production does not follow a circadian rhythm. Furthermore, in these tissues, and also in the pineal gland, melatonin is transformed into its metabolites AFMK and AMK, responsible for antioxidant functions.
Circulating pineal melatonin has a half-life of approximately 30 min in blood and is found free or conjugated with albumin. It is primarily metabolized by the liver, resulting in 6-hydroxymelatonin and, to a lesser extent, N-acetylserotonin. Here, various isoforms of cytochrome P450, specifically enzymes CYP1A1, CYP1A2, CYP1B1, and CYP2C19, produce these metabolites [19]. Then, 6-hydroxymelatonin and N-acetylserotonin are metabolized by sulfotransferases before being excreted in urine [20]. In all tissues, melatonin can be broken down through enzymatic and non-enzymatic processes, producing the metabolites N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK). These metabolites play a crucial role in the elimination of free radicals (Figure 1) [21].
To carry out its multiple functions, melatonin can interact with various receptors or cytosolic proteins, or act independently by itself.
Although it was initially believed that melatonin could cross all biological membranes due to its lipophilic nature [22], it is actually an amphipathic molecule, with hydrophilic and hydrophobic ends. This limits its diffusion capacity through biological membranes. Research in our laboratory has shown that melatonin does not easily cross cell membranes. Instead, it accumulates on the surface of these membranes, and only a small fraction manages to penetrate into the cellular interior, saturating its intranuclear and intramitochondrial levels at certain concentrations [18]. This mechanism is essential to prevent an excess of melatonin inside the cell, as its antioxidant action could interfere with normal energy metabolism.
Melatonin receptors are widely distributed among different tissues in the body, including skeletal muscle. Two types of G-protein-coupled membrane receptors for melatonin have been described in vertebrate animals, MT1 (Mel1a) and MT2 (Mel1b). Both have seven transmembrane domains, and, upon melatonin binding, they can modulate the activity of adenylate cyclase, phospholipases C and A2, potassium and calcium channels, and guanylate cyclase through different mechanisms [23][24]. At one point, the existence of a third melatonin binding site on the cell membrane (MT3 receptor) was theorized [25]. However, it was discovered that this biological target of melatonin was actually the cytosolic enzyme quinone reductase 2 (NQO2) [26]. Other cytosolic proteins, such as calmodulin, calreticulin, tubulin, and protein kinase C, involved in calcium metabolism and cytoskeletal structure modulation, have been shown to be targets of melatonin, exerting various effects upon binding [27][28][29][30].
Melatonin also binds to nuclear receptors, specifically to the subfamily of orphan receptors related to retinoids, which include RORα, RORβ, and RORγ. These receptors are located in the cell nucleus and consist of an N-terminal domain, a DNA binding domain, and a C-terminal domain where they bind to their ligands [31]. Numerous studies demonstrated the presence of melatonin in the cell nucleus, as well as its ability to bind to these receptors, particularly to RORα, regulating gene transcription and carrying out its effects [32][33][34][35][36][37]. Furthermore, like membrane receptors, nuclear receptors exhibit a circadian rhythm similar to that of melatonin, with a peak of production at 3 a.m., suggesting that their expression is regulated by melatonin levels in the blood [38].
Melatonin, along with its metabolites AFMK and AMK, is capable of directly scavenging ROS and RNS [39] (see below).
3. Actions of Pineal and Extrapineal Melatonin
Melatonin is a versatile molecule with the ability to perform multiple functions.
On one hand, pineal melatonin released into circulation reaches every cell in the body and, upon binding to MT1 and MT2 membrane receptors, activates various pathways involved in chronobiological regulation, thus controlling peripheral clocks in different tissues, including skeletal muscle [14][40]. Here, melatonin, through clock genes, jointly coordinates various metabolic, mitochondrial, inflammatory, and structural processes in the muscle following a circadian pattern, as described earlier.
There is evidence that the degradation of clock proteins such as BMAL1, PER, CRY, and REV-ERB, as well as possibly other components, is mediated by the ubiquitin-proteasome system [41][42]. It has been suggested that melatonin may regulate this degradation by inhibiting the proteasome, as it shares similarities with its inhibitor [43][44]. Additionally, it has been observed that melatonin binds to Ca2+/calmodulin [45] and blocks the activity of the Ca2+/calmodulin II-dependent protein kinase (CaMKII) [46], which controls proteasome phosphorylation [47]. This mechanism could be how melatonin regulates clock protein degradation, synchronizing peripheral clocks and providing stability to the rhythm. However, it is currently unknown whether clock genes also control melatonin synthesis in extrapineal tissues [14]. Not in the same way as the in the pineal gland, where it has been demonstrated that the BMAL1:CLOCK complex activates the transcription of AANAT by binding to the E-box region of this gene [48], stimulating rhythmic melatonin production.
While pineal melatonin serves chronobiological functions, extrapineal melatonin acts as an antioxidant and anti-inflammatory agent, with the mitochondria being its primary target [49]. This organelle is clearly affected in aged skeletal muscle.
The antioxidant capacity of melatonin is directly linked to its ability to be produced in various cells of the body in significantly higher quantities than the concentrations of pineal melatonin found in the blood [18], exerting a local effect. Non-circadian expression of genes Aanat and Asmt has been detected in peripheral tissues, such as the heart, liver, stomach, intestines, skeletal muscle, testicles, and ovaries, among others [14][15]. Both melatonin and its metabolites, AFMK and AMK, have the ability to directly neutralize free radicals, interacting with various reactive oxygen and nitrogen species, including hydroxyl radicals (OH•), superoxide radicals (O2−•), hydrogen peroxide (H2O2), nitric oxide (NO), peroxynitrites (ONOO−), and peroxyl radicals (LOO•) [50][51]. Additionally, it has been demonstrated that this hormone is capable of indirectly eliminating free radicals by regulating the activity and expression of other antioxidant systems, an effect that could be mediated by its interaction with calmodulin and its nuclear receptor [52], or with its MT1 and MT2 membrane receptors in the case of pineal melatonin [53]. Firstly, melatonin enhances the activity of antioxidant enzymes such as glutathione peroxidase (GPx) and glutathione reductase (GRd), driving the glutathione cycle and maintaining the balance between oxidized and reduced glutathione (GSSG/GSH) [54][55]. It also stimulates γ-glutamylcysteine synthase, increasing GSH production [56], and glucose-6-phosphate dehydrogenase (G6PD), which provides the necessary NADPH for GRd [57]. Furthermore, melatonin reinforces the activity and expression of other antioxidant enzymes like superoxide dismutase (SOD) and catalase [49][58]. Since the mitochondria are the main source of free radicals, this organelle is its primary target [18]. Beyond its antioxidant function, melatonin plays a crucial role in maintaining mitochondrial homeostasis, preserving the integrity and functionality of membranes, and enhancing mitochondrial bioenergetics [39][59][60][61][62]. These functions of melatonin are especially important in muscle tissue, where mitochondria are critical organelles responsible for regulating the metabolic status of skeletal muscle [63].
Melatonin can also function as an anti-inflammatory molecule by modulating pathways of innate immunity dependent on NF-κB and the NLRP3 inflammasome. Specifically, melatonin binds to its nuclear receptor RORα, activating SIRT1, which in turn deacetylates NF-κB, inhibiting its binding to DNA and its activation, resulting in reduced expression of iNOS and pro-inflammatory cytokines such as TNF-α and IL-6, among others. Thus, melatonin prevents the activation of the NLRP3 inflammasome and caspase-1, as well as IL-1β [64][65]. Additionally, melatonin inhibits the expression of cyclooxygenase-2 (COX-2), preventing excessive production of inflammatory mediators [66]. Furthermore, in stressful situations, melatonin enhances the expression of NRF2, a transcriptional regulator of antioxidant enzymes implicated in maintaining mitochondrial homeostasis [67][68], helping in the anti-inflammatory response. Finally, melatonin also plays a crucial role in apoptosis by inhibiting it through the regulation of the BAX/BCL2 balance and the reduction of caspase-3 activity and expression [69].
All these properties of melatonin make it a potential therapeutic agent against a wide range of age-related diseases, such as sarcopenia, where disruptions in circadian rhythms, chronic inflammation, oxidative stress, mitochondrial damage, and muscle mass loss in aged muscle can be counteracted by melatonin (Figure 2).
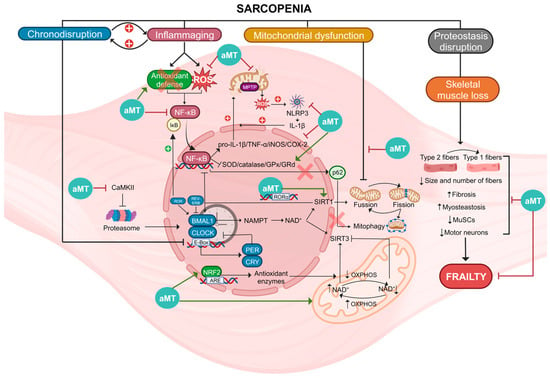
Figure 2. Summary of the molecular pathways involved in sarcopenia and actions of melatonin (aMT). Sarcopenia is a condition associated with several factors, including chronodisruption, inflammaging, mitochondrial dysfunction, proteostasis disruption, and skeletal muscle loss. Chronodisruption leads to the disruption of clock genes and proteins, especially BMAL1, which loses its anti-inflammatory functions. It affects the regulation of NAD+, which is utilized by SIRT1/3, influencing both the NF-κB pathway and OXPHOS. Additionally, the levels of NAD+ are influenced by these two deacetylases. Inflammaging is a process characterized by NF-κB activation caused by elevated ROS levels and diminished antioxidant defenses. This activation results in heightened levels of proinflammatory cytokines and reduced activity of antioxidant enzymes. The oxidative environment causes damage to mitochondria, leading to increased ROS and NLRP3 activation, thereby intensifying inflammation. In this scenario, p62 is unable to initiate mitophagy for the degradation of damaged mitochondria. Mitochondrial dysfunction leads to alterations in fusion, fission, and mitophagy of these organelles. Disruption of proteostasis and the loss of skeletal muscle leads to the switch of type 2 muscle fibers into type 1 and a reduction in both the size and quantity of fibers, MuSCs, and motor neurons. Moreover, there is an increase in fibrosis and myosteatosis. These mechanisms collectively disrupt skeletal muscle integrity, ultimately contributing to frailty. Melatonin, acting at diverse cellular sites, exhibits multiple advantageous properties enabling it to counteract these processes, thereby reducing muscle damage and ameliorating sarcopenia.
4. Melatonin as a Link between Clock Genes and Mitochondria in Sarcopenia
Mitochondria serve as both a source of melatonin synthesis and a target for this indoleamine [18][49]. The decrease in melatonin in tissues associated with aging [70][71] appears to be linked to mitochondrial dysfunction that occurs during this process. Additionally, pineal melatonin production also experiences a significant decline with age [71], potentially leading to reduced circadian control and disruption of clock genes in various tissues. In multiple experimental conditions, including acute and chronic inflammation, aging, and sarcopenia, melatonin consistently enhanced endogenous antioxidant defense, reduced innate immunity activation, and stimulated mitochondria [54][65][72][73][74][75][76][77][78][79][80]. Furthermore, melatonin was also shown to have the capacity to restore clock gene expression, which is altered in these pathologies, as well as in other conditions such as cancer or Parkinson’s [81][82].
Specifically, the research group has demonstrated the benefits of melatonin in aging skeletal muscle. In an initial study led by Sayed et al. [83], using C57BL/6J mice of different ages (3 months, 12 months, and 24 months), the early onset of sarcopenia at 12 months was identified. This was characterized by a decrease in locomotor activity and muscle mass, accompanied by an increase in the frailty index (FI). Additionally, changes in muscle structure and ultrastructure were observed, along with a reduction in the size and loss of type 2 fibers, as well as an increase in mitochondrial size, indicating potential alterations in mitochondrial dynamics. These changes worsened in older animals. Melatonin treatment improved both the function and structure of aged mice’s muscles, while also reducing damage to mitochondria and apoptotic nuclei in the muscle. Therefore, melatonin was suggested as a potential treatment for sarcopenia [78].
Given that sarcopenia is linked to aging, a mouse model lacking NLRP3 was employed to investigate the role of inflammation. It was determined that this inflammasome plays a role in the development and progression of sarcopenia in both skeletal and cardiac muscle. This effect was mitigated with melatonin treatment [74][77][79][80]. Additionally, the expression and circadian rhythm of clock genes were assessed in these mutant mice. Aging resulted in phase changes in Clock, a decrease in the amplitude of Bmal1, Per2, and Clock, and a loss of rhythm in Per2 and Rorα. On the other hand, NLRP3 altered the acrophase of Clock, Per2, and Rorα. Administration of melatonin in these mice allowed for a reduction in age-associated inflammation and restoration of the rhythm of altered genes [73].
Finally, given the growing evidence regarding the importance of the circadian clock in maintaining muscle tissue during aging, particularly in relation to the Bmal1 gene, as detailed in this research, its impact in the gastrocnemius muscle is investigated using an inducible and skeletal-muscle-specific Bmal1 knockout mouse model (iMS-Bmal1−/−) [75]. This aimed to deepen the understanding of the fundamental and underlying mechanisms of sarcopenia, while it is important to note that a mutation in this gene is not considered a genetic model for studying this condition [84]. iMS-Bmal1−/− mice were prone to sarcopenia. These mice experienced changes in their activity/rest rhythms and muscle function, as well as alterations in muscle structure, indicating atrophy, fibrosis, and a shift towards a more oxidative profile in muscle fibers. Additionally, a reduction in mitochondrial oxidative capacity and a decrease in the number of mitochondria, accompanied by damage to them, were observed. Melatonin, through a mechanism that does not require the presence of the Bmal1 gene, was once again able to counteract the changes produced by the absence of this clock gene in these animals.
Overall, these data posit melatonin as a potential candidate against sarcopenia, and suggest that melatonin serves as the link between clock genes and mitochondria in skeletal muscle.
This entry is adapted from the peer-reviewed paper 10.3390/biom13121779
References
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127 (Suppl. 5), 990s–991s.
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646.
- Anker, S.D.; Morley, J.E.; von Haehling, S. Welcome to the ICD-10 code for sarcopenia. J. Cachexia Sarcopenia Muscle 2016, 7, 512–514.
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31.
- Fielding, R.A.; Vellas, B.; Evans, W.J.; Bhasin, S.; Morley, J.E.; Newman, A.B.; Abellan van Kan, G.; Andrieu, S.; Bauer, J.; Breuille, D.; et al. Sarcopenia: An undiagnosed condition in older adults. Current consensus definition: Prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med. Dir. Assoc. 2011, 12, 249–256.
- Cruz-Jentoft, A.J. Sarcopenia, the last organ insufficiency. Eur. Geriatr. Med. 2016, 7, 195–196.
- Ferrucci, L.; de Cabo, R.; Knuth, N.D.; Studenski, S. Of Greek heroes, wiggling worms, mighty mice, and old body builders. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 13–16.
- Morley, J.E.; Abbatecola, A.M.; Argiles, J.M.; Baracos, V.; Bauer, J.; Bhasin, S.; Cederholm, T.; Coats, A.J.; Cummings, S.R.; Evans, W.J.; et al. Sarcopenia with limited mobility: An international consensus. J. Am. Med. Dir. Assoc. 2011, 12, 403–409.
- Scott, D.; Sanders, K.M.; Aitken, D.; Hayes, A.; Ebeling, P.R.; Jones, G. Sarcopenic obesity and dynapenic obesity: 5-year associations with falls risk in middle-aged and older adults. Obesity 2014, 22, 1568–1574.
- Thomas, D.R. Loss of skeletal muscle mass in aging: Examining the relationship of starvation, sarcopenia and cachexia. Clin. Nutr. 2007, 26, 389–399.
- Jeejeebhoy, K.N. Malnutrition, fatigue, frailty, vulnerability, sarcopenia and cachexia: Overlap of clinical features. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 213–219.
- Lerner, A.B.; Case, J.D.; Takahashi, Y. Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands. J. Biol. Chem. 1960, 235, 1992–1997.
- Hardeland, R.; Pandi-Perumal, S.R.; Cardinali, D.P. Melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 313–316.
- Acuña-Castroviejo, D.; Escames, G.; Venegas, C.; Díaz-Casado, M.E.; Lima-Cabello, E.; López, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025.
- Stefulj, J.; Hörtner, M.; Ghosh, M.; Schauenstein, K.; Rinner, I.; Wölfler, A.; Semmler, J.; Liebmann, P.M. Gene expression of the key enzymes of melatonin synthesis in extrapineal tissues of the rat. J. Pineal Res. 2001, 30, 243–247.
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249.
- Liu, T.; Borjigin, J. N-acetyltransferase is not the rate-limiting enzyme of melatonin synthesis at night. J Pineal Res 2005, 39, 91–96.
- Venegas, C.; García, J.A.; Escames, G.; Ortiz, F.; López, A.; Doerrier, C.; García-Corzo, L.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuations. J. Pineal Res. 2012, 52, 217–227.
- Ma, X.; Idle, J.R.; Krausz, K.W.; Gonzalez, F.J. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 2005, 33, 489–494.
- Tian, X.; Huo, X.; Dong, P.; Wu, B.; Wang, X.; Wang, C.; Liu, K.; Ma, X. Sulfation of melatonin: Enzymatic characterization, differences of organs, species and genders, and bioactivity variation. Biochem. Pharmacol. 2015, 94, 282–296.
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42.
- Costa, E.J.; Lopes, R.H.; Lamy-Freund, M.T. Permeability of pure lipid bilayers to melatonin. J Pineal Res 1995, 19, 123–126.
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell. Endocrinol. 2012, 351, 152–166.
- Aranda-Martínez, P.; Fernández-Martínez, J.; Ramírez-Casas, Y.; Guerra-Librero, A.; Rodríguez-Santana, C.; Escames, G.; Acuña-Castroviejo, D. The Zebrafish, an Outstanding Model for Biomedical Research in the Field of Melatonin and Human Diseases. Int. J. Mol. Sci. 2022, 23, 7438.
- Dubocovich, M.L. Melatonin receptors: Are there multiple subtypes? Trends Pharmacol. Sci. 1995, 16, 50–56.
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem. 2000, 275, 31311–31317.
- Benítez-King, G.; Antón-Tay, F. Calmodulin mediates melatonin cytoskeletal effects. Experientia 1993, 49, 635–641.
- Benítez-King, G.; Ríos, A.; Martínez, A.; Antón-Tay, F. In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim. Biophys. Acta 1996, 1290, 191–196.
- Macías, M.; Escames, G.; Leon, J.; Coto, A.; Sbihi, Y.; Osuna, A.; Acuña-Castroviejo, D. Calreticulin-melatonin. An unexpected relationship. Eur. J. Biochem. 2003, 270, 832–840.
- Cardinali, D.P.; Freire, F. Melatonin effects on brain. Interaction with microtubule protein, inhibition of fast axoplasmic flow and induction of crystaloid and tubular formations in the hypothalamus. Mol. Cell. Endocrinol. 1975, 2, 317–330.
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009, 7, e003.
- Menendez-Pelaez, A.; Poeggeler, B.; Reiter, R.J.; Barlow-Walden, L.; Pablos, M.I.; Tan, D.X. Nuclear localization of melatonin in different mammalian tissues: Immunocytochemical and radioimmunoassay evidence. J. Cell. Biochem. 1993, 53, 373–382.
- Acuña-Castroviejo, D.; Pablos, M.I.; Menendez-Pelaez, A.; Reiter, R.J. Melatonin receptors in purified cell nuclei of liver. Res. Commun. Chem. Pathol. Pharmacol. 1993, 82, 253–256.
- Acuña-Castroviejo, D.; Reiter, R.J.; Menéndez-Peláez, A.; Pablos, M.I.; Burgos, A. Characterization of high-affinity melatonin binding sites in purified cell nuclei of rat liver. J. Pineal Res. 1994, 16, 100–112.
- Becker-André, M.; Wiesenberg, I.; Schaeren-Wiemers, N.; André, E.; Missbach, M.; Saurat, J.H.; Carlberg, C. Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J. Biol. Chem. 1994, 269, 28531–28534.
- Wiesenberg, I.; Missbach, M.; Kahlen, J.P.; Schräder, M.; Carlberg, C. Transcriptional activation of the nuclear receptor RZR alpha by the pineal gland hormone melatonin and identification of CGP 52608 as a synthetic ligand. Nucleic Acids Res. 1995, 23, 327–333.
- Carlberg, C.; Wiesenberg, I. The orphan receptor family RZR/ROR, melatonin and 5-lipoxygenase: An unexpected relationship. J. Pineal Res. 1995, 18, 171–178.
- Venegas, C.; García, J.A.; Doerrier, C.; Volt, H.; Escames, G.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Analysis of the daily changes of melatonin receptors in the rat liver. J. Pineal Res. 2013, 54, 313–321.
- Acuña Castroviejo, D.; López, L.C.; Escames, G.; López, A.; García, J.A.; Reiter, R.J. Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem. 2011, 11, 221–240.
- Reiter, R.J. The melatonin rhythm: Both a clock and a calendar. Experientia 1993, 49, 654–664.
- Gatfield, D.; Schibler, U. Physiology. Proteasomes keep the circadian clock ticking. Science 2007, 316, 1135–1136.
- Stojkovic, K.; Wing, S.S.; Cermakian, N. A central role for ubiquitination within a circadian clock protein modification code. Front. Molec. Neurosci. 2014, 7, 69.
- Vriend, J.; Reiter, R.J. Melatonin feedback on clock genes: A theory involving the proteasome. J. Pineal Res. 2015, 58, 1–11.
- Vriend, J.; Reiter, R.J. Melatonin as a proteasome inhibitor. Is there any clinical evidence? Life Sci. 2014, 115, 8–14.
- León, J.; Macías, M.; Escames, G.; Camacho, E.; Khaldy, H.; Martín, M.; Espinosa, A.; Gallo, M.A.; Acuña-Castroviejo, D. Structure-related inhibition of calmodulin-dependent neuronal nitric-oxide synthase activity by melatonin and synthetic kynurenines. Mol. Pharmacol. 2000, 58, 967–975.
- Fukunaga, K.; Horikawa, K.; Shibata, S.; Takeuchi, Y.; Miyamoto, E. Ca2+/calmodulin-dependent protein kinase II-dependent long-term potentiation in the rat suprachiasmatic nucleus and its inhibition by melatonin. J. Neurosci. Res. 2002, 70, 799–807.
- Jarome, T.J.; Kwapis, J.L.; Ruenzel, W.L.; Helmstetter, F.J. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front. Behav. Neurosci. 2013, 7, 115.
- Chong, N.W.; Bernard, M.; Klein, D.C. Characterization of the chicken serotonin N-acetyltransferase gene. Activation via clock gene heterodimer/E box interaction. J. Biol. Chem. 2000, 275, 32991–32998.
- Acuna-Castroviejo, D.; Escames, G.; Rodriguez, M.I.; Lopez, L.C. Melatonin role in the mitochondrial function. Front. Biosci. 2007, 12, 947–963.
- Zhang, H.; Squadrito, G.L.; Uppu, R.; Pryor, W.A. Reaction of peroxynitrite with melatonin: A mechanistic study. Chem. Res. Toxicol. 1999, 12, 526–534.
- Acuña-Castroviejo, D.; Martín, M.; Macías, M.; Escames, G.; León, J.; Khaldy, H.; Reiter, R.J. Melatonin, mitochondria, and cellular bioenergetics. J. Pineal Res. 2001, 30, 65–74.
- Tomás-Zapico, C.; Coto-Montes, A. A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J. Pineal Res. 2005, 39, 99–104.
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolín, I.; Herrera, F.; Martín, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9.
- Escames, G.; López, L.C.; Tapias, V.; Utrilla, P.; Reiter, R.J.; Hitos, A.B.; León, J.; Rodríguez, M.I.; Acuña-Castroviejo, D. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J. Pineal Res. 2006, 40, 71–78.
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000, 14, 1677–1679.
- Urata, Y.; Honma, S.; Goto, S.; Todoroki, S.; Iida, T.; Cho, S.; Honma, K.; Kondo, T. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 27, 838–847.
- Pierrefiche, G.; Laborit, H. Oxygen free radicals, melatonin, and aging. Exp. Gerontol. 1995, 30, 213–227.
- Leon, J.; Acuña-Castroviejo, D.; Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Reiter, R.J. Melatonin and mitochondrial function. Life Sci. 2004, 75, 765–790.
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248.
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357.
- García, J.J.; Piñol-Ripoll, G.; Martínez-Ballarín, E.; Fuentes-Broto, L.; Miana-Mena, F.J.; Venegas, C.; Caballero, B.; Escames, G.; Coto-Montes, A.; Acuña-Castroviejo, D. Melatonin reduces membrane rigidity and oxidative damage in the brain of SAMP8 mice. Neurobiol. Aging 2011, 32, 2045–2054.
- López, A.; García, J.A.; Escames, G.; Venegas, C.; Ortiz, F.; López, L.C.; Acuña-Castroviejo, D. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J. Pineal Res. 2009, 46, 188–198.
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41.
- Acuña-Castroviejo, D.; Rahim, I.; Acuña-Fernández, C.; Fernández-Ortiz, M.; Solera-Marín, J.; Sayed, R.K.A.; Díaz-Casado, M.E.; Rusanova, I.; López, L.C.; Escames, G. Melatonin, clock genes and mitochondria in sepsis. Cell. Mol. Life Sci. 2017, 74, 3965–3987.
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. Faseb J. 2015, 29, 3863–3875.
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524.
- Rahim, I.; Sayed, R.K.; Fernández-Ortiz, M.; Aranda-Martínez, P.; Guerra-Librero, A.; Fernández-Martínez, J.; Rusanova, I.; Escames, G.; Djerdjouri, B.; Acuña-Castroviejo, D. Melatonin alleviates sepsis-induced heart injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Naunyn-Schmiedebergs Arch. Pharmacol. 2021, 394, 261–277.
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88 Pt B, 179–188.
- Mehrzadi, S.; Pourhanifeh, M.H.; Mirzaei, A.; Moradian, F.; Hosseinzadeh, A. An updated review of mechanistic potentials of melatonin against cancer: Pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int. 2021, 21, 188.
- Sanchez-Hidalgo, M.; de la Lastra, C.A.; Carrascosa-Salmoral, M.P.; Naranjo, M.C.; Gomez-Corvera, A.; Caballero, B.; Guerrero, J.M. Age-related changes in melatonin synthesis in rat extrapineal tissues. Exp. Gerontol. 2009, 44, 328–334.
- Hardeland, R. Melatonin in aging and disease -multiple consequences of reduced secretion, options and limits of treatment. Aging Dis. 2012, 3, 194–225.
- Volt, H.; García, J.A.; Doerrier, C.; Díaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205.
- Fernández-Ortiz, M.; Sayed, R.K.A.; Román-Montoya, Y.; de Lama MÁ, R.; Fernández-Martínez, J.; Ramírez-Casas, Y.; Florido-Ruiz, J.; Rusanova, I.; Escames, G.; Acuña-Castroviejo, D. Age and Chronodisruption in Mouse Heart: Effect of the NLRP3 Inflammasome and Melatonin Therapy. Int. J. Mol. Sci. 2022, 23, 6846.
- Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Martínez, J.; Aranda Martínez, P.; Guerra-Librero, A.; Rodríguez-Santana, C.; de Haro, T.; Escames, G.; Acuña-Castroviejo, D.; Rusanova, I. The Impact of Melatonin and NLRP3 Inflammasome on the Expression of microRNAs in Aged Muscle. Antioxidants 2021, 10, 524.
- Fernández-Martínez, J.; Ramírez-Casas, Y.; Aranda-Martínez, P.; López-Rodríguez, A.; Sayed, R.K.A.; Escames, G.; Acuña-Castroviejo, D. iMS-Bmal1(-/-) mice show evident signs of sarcopenia that are counteracted by exercise and melatonin therapies. J. Pineal Res. 2023, e12912.
- Rahim, I.; Djerdjouri, B.; Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Gil, B.; Hidalgo-Gutiérrez, A.; López, L.C.; Escames, G.; Reiter, R.J.; Acuña-Castroviejo, D. Melatonin administration to wild-type mice and nontreated NLRP3 mutant mice share similar inhibition of the inflammatory response during sepsis. J. Pineal Res. 2017, 63, e12410.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Rusanova, I.; Rahim, I.; Escames, G.; López, L.C.; Mokhtar, D.M.; Acuña-Castroviejo, D. The Protective Effect of Melatonin Against Age-Associated, Sarcopenia-Dependent Tubular Aggregate Formation, Lactate Depletion, and Mitochondrial Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1330–1338.
- Sayed, R.K.A.; Fernández-Ortiz, M.; Rahim, I.; Fernández-Martínez, J.; Aranda-Martínez, P.; Rusanova, I.; Martínez-Ruiz, L.; Alsaadawy, R.M.; Escames, G.; Acuña-Castroviejo, D. The Impact of Melatonin Supplementation and NLRP3 Inflammasome Deletion on Age-Accompanied Cardiac Damage. Antioxidants 2021, 10, 1269.
- Fernández-Ortiz, M.; Sayed, R.K.A.; Fernández-Martínez, J.; Cionfrini, A.; Aranda-Martínez, P.; Escames, G.; de Haro, T.; Acuña-Castroviejo, D. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants 2020, 9, 1187.
- Rodríguez-Santana, C.; López-Rodríguez, A.; Martinez-Ruiz, L.; Florido, J.; Cela, O.; Capitanio, N.; Ramírez-Casas, Y.; Acuña-Castroviejo, D.; Escames, G. The Relationship between Clock Genes, Sirtuin 1, and Mitochondrial Activity in Head and Neck Squamous Cell Cancer: Effects of Melatonin Treatment. Int. J. Mol. Sci. 2023, 24, 15030.
- Aranda-Martínez, P.; Fernández-Martínez, J.; Ramírez-Casas, Y.; Rodríguez-Santana, C.; Rusanova, I.; Escames, G.; Acuña-Castroviejo, D. Chronodisruption and Loss of Melatonin Rhythm, Associated with Alterations in Daily Motor Activity and Mitochondrial Dynamics in Parkinsonian Zebrafish, Are Corrected by Melatonin Treatment. Antioxidants 2023, 12, 954.
- Sayed, R.K.; de Leonardis, E.C.; Guerrero-Martínez, J.A.; Rahim, I.; Mokhtar, D.M.; Saleh, A.M.; Abdalla, K.E.; Pozo, M.J.; Escames, G.; López, L.C.; et al. Identification of morphological markers of sarcopenia at early stage of aging in skeletal muscle of mice. Exp. Gerontol. 2016, 83, 22–30.
- Christian, C.J.; Benian, G.M. Animal models of sarcopenia. Aging Cell 2020, 19, e13223.
This entry is offline, you can click here to edit this entry!