p53 is arguably one of the most important tumor suppressor genes in humans. Due to the paramount importance of p53 in the onset of cell cycle arrest and apoptosis, the p53 gene is found either silenced or mutated in the vast majority of cancers. Furthermore, activated wild-type p53 exhibits a strong bystander effect, thereby activating apoptosis in surrounding cells without being physically present there. For these reasons, p53-targeted therapy that is designed to restore the function of wild-type p53 in cancer cells seems to be a very appealing therapeutic approach. Systemic delivery of p53-coding DNA or RNA using nanoparticles proved to be feasible both in vitro and in vivo.
- p53
- gene therapy
- nanoparticles
- bystander effect
- apoptosis
1. Introduction
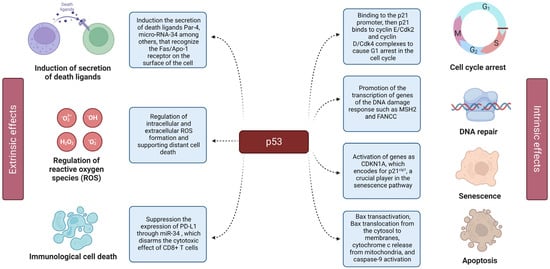
2. p53 in Nanoparticle-Based Gene Therapy for Cancer
2.1. Liposomal Vectors
2.2. Polymer NPs
2.3. Metallic NPs
2.4. Other NPs
3. Impact of NPs on the p53 Protein
| Type of NPs | In Vitro/ In Vivo |
Tissue/Cell Line | Effect | Reference |
|---|---|---|---|---|
| Al2O3 | In vivo | Sub-brain regions of rats | Decreased expression of cyclin D1, bcl-2, Mdm2, and phospho-Rb and increased expression of p53, p21, Bax, and Rb | [43] |
| Ag | In vitro | GC1415, NCI-N87, and MKN45 | Increased p53 expression, inhibition of STAT3 | [44] |
| In vitro | HCT116 | Increased transcription of p53, p21, and caspases (3,8,9), decreased amount of AKT and NF-κB | [45] | |
| CuO | In vitro/ Ex vivo |
K562 and peripheral blood mononuclear cell | Increase in Bax/Bcl-2 ratio, upregulation of p53, and ROS production | [46] |
| Fe3O4 | In vitro | HepG2, A549, IMR-90 | Induction of ROS, upregulation of p53, and caspases 3 and 9 | [47] |
| Pt | In vitro | IMR-90, U251 | Upregulation of p53 and p21, DNA damage | [48] |
| Si | In vitro | HUVECs | Activation of c-Jun, p53, caspase-3, and NF-κB, increased Bax expression and suppression Bcl-2 | [49] |
| TiO2 | Ex vivo | peripheral blood lymphocytes | Accumulation of p53 and activation of DNA damage checkpoint kinases | [50] |
| In vitro | PC12 | ROS and JNK/p53 mediated apoptosis and causing. G2/M arrest by the activation of p53/p21 pathway |
[51] | |
| V2O5 | In vitro | B16F10, A549, and PANC1 | Impaired angiogenesis, increased ROS, overexpression of p53 | [52] |
| Zn | In vitro | HepG2 | ROS generation, DNA damage, activation of p53 and p38 | [53] |
4. Toxicity of NPs
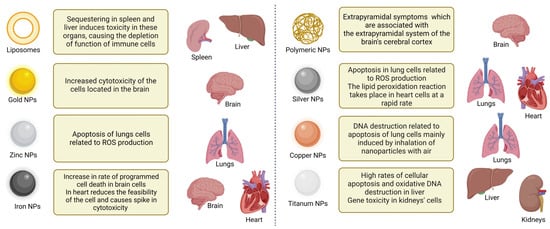
5. Limitations
This entry is adapted from the peer-reviewed paper 10.3390/cells12242803
References
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15–16.
- Rozenberg, J.M.; Zvereva, S.; Dalina, A.; Blatov, I.; Zubarev, I.; Luppov, D.; Bessmertnyi, A.; Romanishin, A.; Alsoulaiman, L.; Kumeiko, V.; et al. The P53 Family Member P73 in the Regulation of Cell Stress Response. Biol. Direct 2021, 16, 23.
- Thomas, A.F.; Kelly, G.L.; Strasser, A. Of the Many Cellular Responses Activated by TP53, which Ones Are Critical for Tumour Suppression? Cell Death Differ. 2022, 29, 961–971.
- Riley, T.; Sontag, E.D.; Chen, P.; Levine, A.J. Transcriptional Control of Human P53-Regulated Genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412.
- Barlev, N.A.; Sayan, B.S.; Candi, E.; Okorokov, A.L. The microRNA and P53 Families Join Forces against Cancer. Cell Death Differ. 2010, 17, 373–375.
- Parfenyev, S.; Singh, A.; Fedorova, O.A.; Daks, A.; Kulshreshtha, R.; Barlev, N.A. Interplay between P53 and Non-Coding RNAs in the Regulation of EMT in Breast Cancer. Cell Death Dis. 2021, 12, 17.
- Hermeking, H. MicroRNAs in the P53 Network: Micromanagement of Tumour Suppression. Nat. Rev. Cancer 2012, 12, 613–626.
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A.; Barlev, N.A. Lysine-Specific Modifications of P53: A Matter of Life and Death? Oncotarget 2013, 4, 1556–1571.
- Liu, Y.; Tavana, O.; Gu, W. P53 Modifications: Exquisite Decorations of the Powerful Guardian. J. Mol. Cell Biol. 2019, 11, 564–577.
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 Inhibition: An Important Step Forward in Cancer Therapy. Leukemia 2020, 34, 2858–2874.
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The Roles and Regulation of MDM2 and MDMX: It Is Not Just about P53. Genes Dev. 2021, 35, 575–601.
- Morgunkova, A.; Barlev, N.A. Lysine Methylation Goes Global. Cell Cycle 2006, 5, 1308–1312.
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 Affects Genotoxic Stress Response via the Mdm2 Axis. Oncotarget 2015, 6, 25843–25855.
- Petukhov, A.; Ag, M.; Moiseeva, T.N.; Tn, M.; Barlev, N.A. Role of Proteasomes in Transcription and Their Regulation by Covalent Modifications. Front. Biosci. 2008, 13, 7184–7192.
- Sdek, P.; Ying, H.; Chang, D.L.F.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Allday, M.J.; Xiao, Z.-X.J. MDM2 Promotes Proteasome-Dependent Ubiquitin-Independent Degradation of Retinoblastoma Protein. Mol. Cell 2005, 20, 699–708.
- Stindt, M.H.; Carter, S.A.; Vigneron, A.M.; Ryan, K.M.; Vousden, K.H. MDM2 Promotes SUMO-2/3 Modification of P53 to Modulate Transcriptional Activity. Cell Cycle 2011, 10, 3176–3188.
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W.; Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 Promotes the Neddylation of P53 and Inhibits Its Transcriptional Activity. J. Biol. Chem. 2007, 282, 1797–1804.
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a Activates P53 through Methylation-Independent Mechanism. Oncogene 2017, 36, 922–932.
- Daks, A.; Shuvalov, O.; Fedorova, O.; Parfenyev, S.; Simon, H.-U.; Barlev, N.A. Barlev Methyltransferase Set7/9 as a Multifaceted Regulator of ROS Response. Int. J. Biol. Sci. 2023, 19, 2304–2318.
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F.; Reinberg, D.; Barlev, N.A. Methylation-Acetylation Interplay Activates P53 in Response to DNA Damage. Mol. Cell. Biol. 2007, 27, 6756–6769.
- Xu, M.; Kumar, D.; Kumar, D.; Kumar, D.; Srinivas, S.; DeTolla, L.J.; Yu, S.F.; Stass, S.A.; Mixson, A.J. Parenteral Gene Therapy with P53 Inhibits Human Breast Tumors in Vivo through a Bystander Mechanism without Evidence of Toxicity. Hum. Gene Ther. 1997, 8, 177–185.
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of P53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054.
- Brady, C.A.; Attardi, L.D. P53 at a Glance. J. Cell Sci. 2010, 123, 2527–2532.
- Bellazzo, A.; Sicari, D.; Valentino, E.; Del Sal, G.; Collavin, L. Complexes Formed by Mutant P53 and Their Roles in Breast Cancer. Breast Cancer 2018, 10, 101–112.
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting Mutant P53 for Efficient Cancer Therapy. Nat. Rev. Cancer 2018, 18, 89–102.
- Vassilev, L.T. MDM2 Inhibitors for Cancer Therapy. Trends Mol. Med. 2007, 13, 23–31.
- Davidovich, P.; Aksenova, V.; Petrova, V.; Tentler, D.; Orlova, D.; Smirnov, S.; Gurzhiy, V.; Okorokov, A.L.; Garabadzhiu, A.; Melino, G.; et al. Discovery of Novel Isatin-Based P53 Inducers. ACS Med. Chem. Lett. 2015, 6, 856–860.
- Fallatah, M.M.J.; Law, F.V.; Chow, W.A.; Kaiser, P. Small-Molecule Correctors and Stabilizers to Target P53. Trends Pharmacol. Sci. 2023, 44, 274–289.
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.I.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-Human Study of PC14586, a Small Molecule Structural Corrector of Y220C Mutant P53, in Patients with Advanced Solid Tumors Harboring a TP53 Y220C Mutation. J. Clin. Oncol. 2022, 40 (Suppl. S16), 3003.
- Gounder, M.; Bauer, T.; Schwartz, G.; Weise, A.; LoRusso, P.; Kumar, P.; Tao, B.; Hong, Y.; Patel, P.; Lu, Y.; et al. A First-in-Human Phase I Study of Milademetan, an MDM2 Inhibitor, in Patients With Advanced Liposarcoma, Solid Tumors, or Lymphomas. J. Clin. Oncol. 2023, 41, 1714–1724.
- Mahfoudhi, E.; Lordier, L.; Marty, C.; Pan, J.; Roy, A.; Roy, L.; Rameau, P.; Abbes, S.; Debili, N.; Raslova, H.; et al. P53 Activation Inhibits All Types of Hematopoietic Progenitors and All Stages of Megakaryopoiesis. Oncotarget 2016, 7, 31980–31992.
- Khurana, A.; Shafer, D.A. MDM2 Antagonists as a Novel Treatment Option for Acute Myeloid Leukemia: Perspectives on the Therapeutic Potential of Idasanutlin (RG7388). OncoTargets Ther. 2019, 12, 2903–2910.
- Daks, A.; Petukhov, A.; Fedorova, O.; Shuvalov, O.; Merkulov, V.; Vasileva, E.; Antonov, A.; Barlev, N.A. E3 Ubiquitin Ligase Pirh2 Enhances Tumorigenic Properties of Human Non-Small Cell Lung Carcinoma Cells. Genes Cancer 2016, 7, 383–393.
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698.
- Zeimet, A.G.; Marth, C. Why Did P53 Gene Therapy Fail in Ovarian Cancer. Lancet Oncol. 2003, 4, 415–422.
- Szewczyk, O.K.; Roszczenko, P.; Czarnomysy, R.; Bielawska, A.; Bielawski, K. An Overview of the Importance of Transition-Metal Nanoparticles in Cancer Research. Int. J. Mol. Sci. 2022, 23, 6688.
- Rezaei, M.; Esmailzadeh, A.; Shanei, A. Bystander Effect of Therapeutic Ultrasound in the Presence of Cisplatin: An in Vitro Study on Human Melanoma Cells. J. Biomed. Phys. Eng. 2023, 13, 433–442.
- He, W.; Yan, J.; Li, Y.; Yan, S.; Wang, S.; Hou, P.; Lu, W. Resurrecting a P53 Peptide Activator—An Enabling Nanoengineering Strategy for Peptide Therapeutics. J. Control. Release 2020, 325, 293–303.
- Kim, C.-K.; Choi, E.J.; Choi, E.-J.; Choi, S.-H.; Park, J.-S.; Haider, K.H.; Ahn, W.S. Enhanced P53 Gene Transfer to Human Ovarian Cancer Cells Using the Cationic Nonviral Vector, DDC. Gynecol. Oncol. 2003, 90, 265–272.
- Marvalim, C.; Datta, A.; Lee, S.C. Role of P53 in Breast Cancer Progression: An Insight into P53 Targeted Therapy. Theranostics 2023, 13, 1421–1442.
- Prabha, S.; Labhasetwar, V. Nanoparticle-Mediated Wild-Type P53 Gene Delivery Results in Sustained Antiproliferative Activity in Breast Cancer Cells. Mol. Pharm. 2004, 1, 211–219.
- Kotcherlakota, R.; Vydiam, K.; Srinivasan, D.J.; Mukherjee, S.; Roy, A.; Kuncha, M.; Rao, T.N.; Sistla, R.; Gopal, V.; Patra, C.R. Restoration of P53 Function in Ovarian Cancer Mediated by Gold Nanoparticle-Based EGFR Targeted Gene Delivery System. ACS Biomater. Sci. Eng. 2019, 5, 3631–3644.
- Liu, H.; Zhang, W.; Fang, Y.; Yang, H.; Tian, L.; Li, K.; Lai, W.; Bian, L.; Lin, B.; Liu, X.; et al. Neurotoxicity of Aluminum Oxide Nanoparticles and Their Mechanistic Role in Dopaminergic Neuron Injury Involving P53-Related Pathways. J. Hazard. Mater. 2020, 392, 122312.
- Huang, D.; Wang, J.; Zhou, S.; Zhang, T.; Cai, J.; Liu, Y. Ag Nanoparticles Green-Mediated by Scrophularia Striata Aqueous Extract Induce Apoptosis via P53 and Signal Transducer and Activator of Transcription 3 Signaling Pathways in Gastric Cancer Cells. Inorg. Chem. Commun. 2023, 155, 110942.
- Satapathy, S.R.; Mohapatra, P.; Preet, R.; Das, D.; Sarkar, B.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Silver-Based Nanoparticles Induce Apoptosis in Human Colon Cancer Cells Mediated through P53. Nanomed. Nanotechnol. Biol. Med. 2013, 8, 1307–1322.
- Shafagh, M.; Rahmani, F.; Delirezh, N. CuO nanoparticles induce cytotoxicity and apoptosis in human K562 cancer cell line via mitochondrial pathway, through reactive oxygen species and P53. Iran. J. Basic Med. Sci. 2015, 18, 993–1000.
- Ahamed, M.; Alhadlaq, H.A.; Khan, M.A.M.; Akhtar, M.J. Selective Killing of Cancer Cells by Iron Oxide Nanoparticles Mediated through Reactive Oxygen Species via P53 Pathway. J. Nanopart. Res. 2013, 15, 1225.
- Asharani, P.V.; Xinyi, N.; Hande, M.P.; Valiyaveettil, S. DNA Damage and P53-Mediated Growth Arrest in Human Cells Treated with Platinum Nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2010, 5, 51–64.
- Liu, X.; Sun, J. Endothelial Cells Dysfunction Induced by Silica Nanoparticles through Oxidative Stress via JNK/P53 and NF-κB Pathways. Biomaterials 2010, 31, 8198–8209.
- Kang, S.J.; Kim, B.M.; Lee, Y.-J.; Chung, H.W. Titanium Dioxide Nanoparticles Trigger P53-Mediated Damage Response in Peripheral Blood Lymphocytes. Environ. Mol. Mutagen. 2008, 49, 399–405.
- Wu, J.; Sun, J.; Xue, Y. Involvement of JNK and P53 Activation in G2/M Cell Cycle Arrest and Apoptosis Induced by Titanium Dioxide Nanoparticles in Neuron Cells. Toxicol. Lett. 2010, 199, 269–276.
- Xi, W.; Tang, H.; Liu, Y.; Liu, C.; Gao, Y.; Cao, A.; Liu, Y.; Chen, Z.; Wang, H. Cytotoxicity of Vanadium Oxide Nanoparticles and Titanium Dioxide-coated Vanadium Oxide Nanoparticles to Human Lung Cells. J. Appl. Toxicol. 2020, 40, 567–577.
- Sharma, V.; Anderson, D.; Kumar, A.; Dhawan, A. Zinc Oxide Nanoparticles Induce Oxidative DNA Damage and ROS-Triggered Mitochondria Mediated Apoptosis in Human Liver Cells (HepG2). Apoptosis 2012, 17, 852–870.
- Roszczenko, P.; Szewczyk, O.K.; Czarnomysy, R.; Bielawski, K.; Bielawska, A. Biosynthesized Gold, Silver, Palladium, Platinum, Copper, and Other Transition Metal Nanoparticles. Pharmaceutics 2022, 14, 2286.
- Yao, Y.; Zang, Y.; Qu, J.; Tang, M.; Zhang, T. The Toxicity Of Metallic Nanoparticles On Liver: The Subcellular Damages, Mechanisms, And Outcomes. Int. J. Nanomed. 2019, 14, 8787–8804.
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.-C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190.
- Dolma, L.; Muller, P.A.J. GOF Mutant P53 in Cancers: A Therapeutic Challenge. Cancers 2022, 14, 5091.
- Munisamy, M.; Mukherjee, N.; Thomas, L.; Pham, A.T.; Shakeri, A.; Zhao, Y.; Kolesar, J.; Rao, P.P.N.; Rangnekar, V.M.; Rao, M. Therapeutic Opportunities in Cancer Therapy: Targeting the P53-MDM2/MDMX Interactions. Am. J. Cancer Res. 2021, 11, 5762–5781.
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-Function Mutant P53 in Cancer Progression and Therapy. J. Mol. Cell Biol. 2020, 12, 674–687.
- Matissek, K.J.; Mossalam, M.; Okal, A.; Lim, C.S. The DNA Binding Domain of P53 Is Sufficient To Trigger a Potent Apoptotic Response at the Mitochondria. Mol. Pharm. 2013, 10, 3592–3602.
- Okal, A.; Matissek, K.J.; Matissek, S.J.; Price, R.; Salama, M.E.; Janát-Amsbury, M.M.; Lim, C.S. Re-Engineered P53 Activates Apoptosis in Vivo and Causes Primary Tumor Regression in a Dominant Negative Breast Cancer Xenograft Model. Gene Ther. 2014, 21, 903–912.
- Matissek, K.J.; Okal, A.; Mossalam, M.; Lim, C.S. Delivery of a Monomeric P53 Subdomain with Mitochondrial Targeting Signals from Pro-Apoptotic Bak or Bax. Pharm. Res. 2014, 31, 2503–2515.
- Lu, P.; Redd Bowman, K.E.; Brown, S.M.; Joklik-Mcleod, M.; Vander Mause, E.R.; Nguyen, H.T.N.; Lim, C.S. P53-Bad: A Novel Tumor Suppressor/Proapoptotic Factor Hybrid Directed to the Mitochondria for Ovarian Cancer Gene Therapy. Mol. Pharm. 2019, 16, 3386–3398.
- Waterman, M.J.; Waterman, J.L.; Halazonetis, T.D. An Engineered Four-Stranded Coiled Coil Substitutes for the Tetramerization Domain of Wild-Type P53 and Alleviates Transdominant Inhibition by Tumor-Derived P53 Mutants. Cancer Res. 1996, 56, 158–163.