Antisense oligonucleotide-based (ASO) therapeutics have emerged as a promising strategy for the treatment of human disorders. Charge-neutral PMOs have promising biological and pharmacological properties for antisense applications. Despite their great potential, the efficient delivery of these therapeutic agents to target cells remains a major obstacle to their widespread use. Cellular uptake of naked PMO is poor. Cell-penetrating peptides (CPPs) appear as a possibility to increase the cellular uptake and intracellular delivery of oligonucleotide-based drugs. Among these, the DG9 peptide has been identified as a versatile CPP with remarkable potential for enhancing the delivery of ASO-based therapeutics due to its unique structural features. Notably, in the context of phosphorodiamidate morpholino oligomers (PMOs), DG9 has shown promise in enhancing delivery while maintaining a favorable toxicity profile.
1. Introduction
The advancement of antisense oligonucleotides (ASOs) has brought about a profound change in the field of genetic therapeutics, offering a promising avenue for addressing a diverse array of diseases on a molecular level. ASOs are short synthetic nucleic acid analogs that offer a revolutionary means to modulate gene expression by precisely interacting with RNA transcripts. The history of ASO can be traced back to the pioneering work of Zamecnik and Stephenson in early 1970, who first proposed the concept of using synthetic oligonucleotides to regulate eukaryotic gene expression in cultured cells through sequence-specific hybridization with RNA [
1,
2]. Later, the pharmacokinetic properties of ASOs, such as stability, reduced susceptibility to nuclease degradation, specificity, and cellular absorption, have been greatly improved by developments in oligonucleotide chemistry, including the introduction of chemical modifications and different backbone structures, which transformed them from theoretical concepts into potentially effective therapeutic agents [
3].
ASOs have been successfully employed in treating a wide range of diseases, including Duchenne Muscular Dystrophy (DMD), spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and many more. This success led to the regulatory approval of 10 ASO-based drugs [
4] and many antisense drug candidates for clinical trials to treat cardiovascular, metabolic, endocrine, neurological, neuromuscular, inflammatory, and infectious diseases [
5]. This demonstrates the dynamic nature of ASO-mediated therapy. Despite being a promising approach, it is widely accepted that the delivery of ASO treatments to specific tissues is limited by factors such as intracellular trafficking, degradation in biological fluids, and transportation across cellular barriers [
6]. Although chemical modifications have significantly improved their metabolic stability as well as their affinities for RNA targets and have, to some extent, reduced off-target effects, no chemical modification has significantly improved cellular uptake or tissue targeting.
Cell-penetrating peptides (CPPs) or peptide transduction domains (PTDs) are one of the many approaches that have been developed to improve the delivery of oligonucleotides. CPPs are small peptides with the ability to transport cargos, including ASOs, across cellular barriers and hereby offer the potential to improve ASOs’ cellular uptake and intracellular distribution, enhancing therapeutic outcomes and reducing the required dosage [
7]. The initial CPP was introduced several decades ago, and ever since, there has been an ongoing endeavor to enhance cell-penetrating peptides for improved oligonucleotide delivery and enhanced pharmacological properties [
8].
Particularly in the context of phosphorodiamidate morpholino oligomers (PMOs), R6G, PiP (PNA/PMO Internalizing Peptides), and DG9 have captured interest among the CPPs for their potential to improve ASO-mediated therapy. PMOs have shown effectiveness in treating genetic diseases, but their poor cellular absorption continues to be a major drawback. Due to its high efficacy and low toxicity, DG9 has become a promising CPP for improving the intracellular transport of PMOs since it holds the prospect of improved therapeutic advantages [
9,
10].
2. The Mode of Action of Antisense Oligonucleotide
Antisense oligonucleotides (ASOs) are synthetic, single-stranded nucleic acid molecules targeted for mRNA, generally comprised of ~18–30 nucleotides with a variety of chemical structures [
12]. ASOs form a DNA–RNA hybrid by binding specific RNA sequences through Watson–Crick base pairing to modulate gene expression [
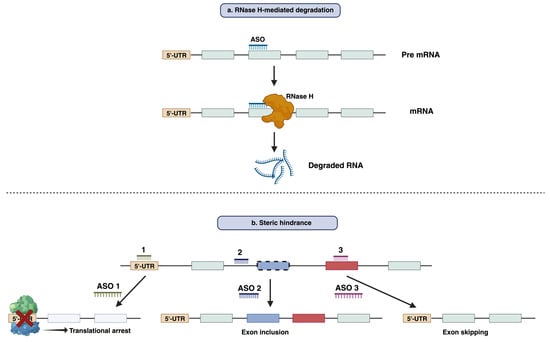
13]. The functional mechanism of ASO can be broadly categorized into two main modes of action: RNase H-mediated degradation and steric hindrance [
13].
RNase H-mediated degradation: When DNA-based oligonucleotides, also known as gapmers, bind to their respective mRNA sequences, they can recruit endogenous RNase H enzymes. RNase H recognizes the RNA–DNA duplex and catalyzes the degradation of RNA, leading to the reduction in the target RNA and gene silencing (
Figure 1a) [
14]. This strategy has been employed widely to suppress disease-causing or disease-modifying genes. Fomivirsen, mipomersen, and inotersen are the three RNase H-competent ASOs that have so far acquired regulatory approval [
14].
Figure 1. Functional mechanism of antisense oligonucleotide-mediated modulation of gene expression. (a) RNase H mediated degradation of RNA by antisense oligonucleotides. (b) Suppressing the translation or splicing modulation by an antisense oligonucleotide through steric hindrance mechanisms.
Steric hindrance: Apart from RNase H-mediated breakdown, ASOs can interfere with RNA–RNA or RNA–Protein interaction by blocking certain regions within the target transcript. This results in the prevention of translation rather than the lowering of transcript levels [
15]. The best-known application of this mode of action is splicing modulation, which can cause either exclusion (exon skipping) or retention (exon inclusion) of specific exon/exons by targeting splice sites or exonic/intronic inclusion signals, respectively [
16,
17]. Typically, this approach can be used both for restoration of the translational reading frame to have functional protein synthesis or for disruption of translation of the target gene [
18,
19] (
Figure 1b). Eteplirsen, golodirsen, nusinersen, viltolarsen, casimersen, milasen, and atipeksen are the splice-switching ASOs that have received FDA approval to date [
12,
20,
21,
22].
3. Molecular Mechanism of Cellular Uptake and Intracellular Distribution of Antisense Oligonucleotides
The effectiveness of antisense oligonucleotides (ASOs) as therapeutic agents depends significantly on cellular uptake and intracellular distribution. To have the desired effects, ASOs must efficiently penetrate cells and locate their target locations. After intravenous, subcutaneous, or direct administration, ASOs reach the bloodstream, where they can be broken down by nucleases [
23]. Once they reach the target organ, the cellular uptake process can be achieved in several ways, such as phagocytosis, macropinocytosis, micropinocytosis via clathrin and caveolin-independent pathways, caveolar internalization, and classical clathrin-mediated endocytosis. Following cellular uptake, ASOs are internalized into early endosomes and then late endosomes, regulated by Rab, SNARE, and tethering proteins. A percentage of ASO drugs, possibly a very tiny portion, are released from late endosomes into the cytoplasm, where they target mRNAs or pre-mRNAs in the cytoplasm or the nucleus to carry out their therapeutic effects. Nuclear entry can be actively mediated by the nuclear pore mechanism or passively via simple diffusion [
24]. Many small cellular proteins, such as COPII, can facilitate nuclear trafficking. However, the process is not entirely known [
23]. The target of different ASOs is located at different subcellular sites. For RNase H-mediated mRNA degradation, the ASO drugs need to reach either the cytoplasm (ribosomes) or the nucleus [
25]. In contrast, for exon skipping/inclusion, ASOs must be present in the spliceosomes of the nucleus [
26].
4. Challenges Associated with ASO Delivery
Although ASOs have great potential as therapeutic agents, their efficient delivery faces several difficulties. These difficulties are associated with the physiochemical characteristics of ASO molecules, such as their large size, molecular weight (single-stranded ASOs are ~4–10 kDa, double-stranded siRNAs are ~14 kDa), and negative charge, which hinders passive diffusion across the cell membrane. ASOs predominantly rely on endocytosis for cellular uptake, which might be ineffective and lead to entrapment in endosomes or lysosomes, leading to lysosomal degradation. So, once inside the cell, ASO must escape endosomal entrapment to gain access to the target region in the cytoplasm or nucleus [
27]. Apart from that, for the systemically administered ASOs to be effective, they need to avoid renal clearance [
28,
29], resist nuclease degradation both in the extracellular fluid and intracellular compartment [
30], and avoid removal by the reticuloendothelial system, which includes mononuclear phagocytes, liver sinusoidal endothelial cells, and Kupffer cells [
31]. A study reported that intravenous administration of an AON resulted in 40% and 18% accumulation in the liver and kidneys, respectively [
32].
Due to these challenges, to date, most of the approved oligonucleotide treatments are delivered either locally (for example, to the eye or spinal cord) or to the liver. The eye is chosen as a target for ASO delivery (for example, Pegaptanib and Fomivirsen) due to its accessibility, anatomical considerations, and immune-privileged status [
12]. Although ocular delivery of ASOs has benefits, there are still obstacles to be overcome, including getting through anatomical obstacles (such as the blood–retinal barrier), maximizing ASO stability, and pharmacokinetics for long-lasting therapeutic effects. For ASOs targeting the CNS, direct delivery into the cerebrospinal fluid via lumber puncture is most commonly used (for example, Nusinersen) [
34]. However, it should be noted that this method requires expertise and specialized equipment and carries a small risk of complications associated with invasive procedures.
5. Strategies to Enhance the Stability and Delivery of Antisense Oligonucleotides
5.1. Chemical Modification
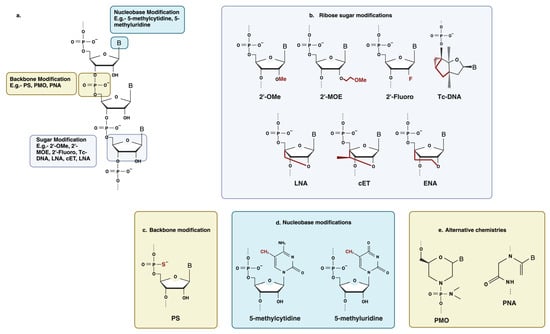
Antisense oligonucleotides were initially employed as synthesized, unaltered DNA, which turned out to be extremely vulnerable to exonuclease and endonuclease degradation [
35] (
Figure 3). Chemical modifications of antisense oligonucleotides can enhance stability, improve target binding affinity and biodistribution, and provide protection against nuclease-mediated degradation. Modification of the nucleic acid backbone, the ribose sugar moiety, and the nucleobase itself have been extensively employed to improve the drug-like properties of antisense oligonucleotides [
29,
36].
Figure 3. Some common chemical modifications used in antisense oligonucleotide chemistry. (
a) Schematic of an RNA nucleotide with a common modification site. (
b) Ribose sugar modification: 2′-OMe, 2′-O-methyl; 2′-MOE, 2′-O-methoxyethyl; 2′-Fluoro; tcDNA, tricyclo DNA; LNA, locked nucleic acid; cEt, constrained ethyl bridged nucleic acid; ENA, ethylene-bridged nucleic acid. (
c) Backbone modification: PS, phosphorothioate. (
d) Nucleobase modification: 5-methylcytidine, 5-methyluridine. (
e) Alternative chemistries: PMO, phosphorodiamidate morpholino oligonucleotide; PNA, peptide nucleic acid. Created with BioRender.com (
https://app.biorender.com/illustrations/64c764c0257fb4bbb5688afa (Accessed on 2 August 2023).
5.2. Bioconjugates
While chemical modifications are required to protect ASOs from exonucleases and prolong their stability, the next challenge is ASO passage across biological barriers. These barriers include the vascular endothelial barrier, cell membranes, and intracellular compartments. Additionally, achieving specific cell/tissue targeting and a reduction in clearance from circulation is essential [
85]. Improving ASO delivery potential can be achieved through the conjugation of different moieties that can direct the drug to specific tissues and enhance internalization. Bioconjugates are distinct molecular entities with precise stoichiometry, which ensures well-defined pharmacokinetic properties and simplifies large-scale synthesis. Additionally, bioconjugates tend to have a small size, which often results in favorable biodistribution profiles [
12]. Bioconjugates usually promote interaction with cell-type-associated receptors, consequently enhancing delivery to the target tissue and internalization by receptor-mediated endocytosis [
86]. There are different types of conjugates available, including lipid-based bioconjugates (e.g., cholesterol and its derivatives) [
87,
88,
89], peptide-based bioconjugates (e.g., cell-penetrating peptides) [
90,
91,
92,
93,
94,
95], aptamers [
96], antibodies [
97,
98], sugars (for example, N-acetylgalactosamine (GalNAc)) [
99,
100], and polymers (e.g., PEG) (
Table 2). The selection of the appropriate bioconjugate depends on several factors, including the application goals, specific requirements of the ASO delivery system, the intended therapeutic application, and safety considerations. Due to the effectiveness of bioconjugates in increasing the efficacy of ASO delivery, bioconjugated compounds are present in four of the five FDA-approved siRNA medications [
101].
Table 2. Brief description of the most commonly used bioconjugates in the delivery of antisense oligonucleotides.
| Bioconjugates |
Brief Introduction |
Benefits |
| Lipid-based conjugates |
Lipid-based moieties are usually cholesterol and its derivatives, which are covalently conjugated to siRNA and antagomir ASOs to enhance delivery. This group of bioconjugates enhances in vivo delivery by adhering to lipoprotein particles (such as HDL and LDL) in the circulation and taking over the body’s natural system for lipid uptake and transport [101]. The overall hydrophobicity of siRNAs governs their in vivo association with the various classes of lipoprotein, with the more hydrophobic conjugates preferentially attaching to LDL and primarily being taken up by the liver. The less lipophilic conjugates preferentially bind to HDL and are consumed by the liver, adrenal glands, ovary, kidney, and small intestine. Another lipid derivative, α-tocopherol (vitamin E), was also found to increase the delivery of siRNA [12]. |
-
Improved cellular uptake.
-
Enhanced pharmacokinetic properties.
-
Improved cell/tissue targeting.
-
Enhanced binding specificity.
-
Improved in vivo stability.
|
| GalNac conjugates |
Trimeric GalNac is the most clinically successful tissue-targeting ligand used in ASO delivery to date. GalNAc is a carbohydrate moiety that has a high affinity for the highly expressed asialoglycoprotein receptor 1 (ASGR1 and ASPGR) [101]. This interaction promotes the endocytosis of PO ASOs and siRNAs into hepatocytes. Givosiran, a GalNAc-conjugated siRNA, was granted FDA approval for the treatment of acute hepatic porphyria in November 2019 as a result of its remarkable success [12]. |
| Antibody and Aptamer conjugates |
Antibody–RNA bioconjugates offer a promising strategy for nucleic acid therapeutics; however, their utility for oligonucleotide delivery is still in the early stages of development. Antibodies are useful for the targeted delivery of oligonucleotides to cells or tissues that other methods cannot reach since they are very selective in recognizing target antigens [12,101]. Aptamers bind to their specific target proteins with high affinity, just like antibodies do. Aptamers are regarded as chemical antibodies and have demonstrated many advantages over antibodies, including being easier and less expensive to produce (i.e., through chemical synthesis), smaller size, and lower immunogenicity [12]. |
| Polymer conjugates |
PEG is a non-ionic, hydrophilic polymer with a wide range of applications. It is widely used to prolong blood circulation and improve drug efficacy. PEGylation, which involves covalently adding PEG to a drug, improves the stability of ASOs and reduces renal excretion by forming a protective hydration layer around them. PEG-conjugated drugs have been found to have better pharmacokinetic and pharmacodynamic properties in terms of the drug’s chemical aspects of absorption, distribution, metabolism, excretion, and toxicity (ADMET). Other polymers besides PEG have also received attention, including poly(glycerol), poly(2-oxazoline), poly (amino acid), and poly[N-(2-hydroxypropyl)methacrylamide] because they are more ADMET-enhancing and less immunogenic [101]. |
| Peptide-based conjugates |
Peptides are short chains of amino acids that can serve as carriers for oligonucleotide delivery for their cell-specific targeting, cell-penetrating, or endosomolytic properties [12]. |
7. Overcoming the Limitations of PMO by Conjugating It with Cell Penetrating Peptides
PMOs show low efficacy as therapeutic agents due to their poor cellular uptake, less permeability of membrane barriers, rapid clearance from the systemic circulation, inability to cross blood–brain barriers, and the requirement of repetitive administration and/or a high dosage of the drug for executing its function. Apart from that, due to the hydrophobicity of the plasma membrane and the neutral charge in PMO, only small portions of internalized PMOs can escape endosomes and reach their intended target [
83]. A promising utilization of CPP is their ability to directly conjugate with neutrally charged PMO and PNA and increase the delivery efficacy [
109,
110,
111].
Conjugation of CPPs with PMO is one such approach to improving PMO delivery. This strategy was first demonstrated with an arginine-rich peptide (RXR)4, which was administered to the
mdx mouse model of DMD in a variety of doses, time intervals, and delivery methods. It was observed that a single intravenous administration can cause high dystrophin exon skipping in skeletal muscle, the diaphragm, and for the first time in the heart [
116]. Another arginine-rich peptide, (RXRRBR)2 peptide (B-peptide), was identified from a screen using the EGFP-654 splicing reporter mouse model to ensure PPMO entry to cells and notable exon-skipping in the heart after retro-orbital injection, resulting in improved cardiac function, specifically end-systolic volume and end-diastolic volume and resistance to dobutamine [
117]. In another study, intravenous injection of a single 25 mg/kg dose of B-peptide conjugated PMO into
mdx mice confirmed approximately 50% of wild-type dystrophin levels along with restoration in cardiac function [
116,
117] and improved muscle function. In contrast, weekly administration of naked PMO at 200 mg/kg for 12 weeks could only achieve 10% wild-type dystrophin levels [
118]. Fusion of muscle-specific peptide (MSP) with B-peptide through a phage display has been found to improve activity 2- to 4-fold after multiple 6 mg/kg doses [
119].
Recent research has led to the development of several peptide series known as “Pip’s” (PMO/PNA internalization peptides), which are generated from the parent peptide penetratin [
125,
126] and consist of the amino acids arginine (R), 6-aminohexanoic acid (X), and ß-alanine spacer (B), with an internal core containing hydrophobic residues [
12]. The most recent Pip-PMO conjugates are significantly more effective than naked PMO and, more critically, reach cardiac muscle following systemic administration in dystrophic animal models. A single intravenous injection of the Pip5e peptide-conjugated PMO induced the highest amounts of exon skipping and dystrophin restoration throughout the body, including in the hearts of
mdx mice [
127]. To increase homogenous dystrophin repair and to target the heart muscle more effectively, the Pip6 series of peptides were generated by further iterations of the core design [
128].
CPP-PMO has also been used as a therapeutic approach for myotonic dystrophy type I, where a CTG expansion in the DMPK gene’s 3′ untranslated region causes a pathogenic transcript that interacts with RNA-binding proteins like muscleblind-like 1 (MBNL1) to cause widespread aberrant splicing abnormalities. Systemic administration of B-PMO targeting this repeat element causes the blocking of Mbnl1 sequestration, resulting in normal nuclear distribution and subsequent correction of abnormal RNA splicing, including for the chloride channel 1 gene, which is a primary contributor to myotonia [
131].
One of the biggest challenges of nucleic acid therapy is crossing the blood–brain barrier to reach the central nervous system (CNS) after systemic delivery. CPPs have been identified as promising medicines in the treatment of central nervous system (CNS) diseases due to their demonstrated transmembrane transporting ability. It is assumed that small-size cationic or amphipathic CPPs may exhibit greater affinity for negatively charged endothelial cells on the blood–brain barrier [
132,
133]. CPP-PMOs have recently been investigated in preclinical models of spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disorder that results in premature death [
134]. This disease is caused by mutations in the survival of the motor neuron 1 (SMN1) gene. A paralogous gene, SMN2, encodes a vital SMN protein but generates only minimal levels due to a sequence variant leading to the exclusion of exon 7 from approximately 90% of mature transcripts. Consequently, a truncated, non-functional protein is produced [
135,
136]. To address the functional deficiency caused by the loss of SMN1 protein in patients, ASOs have been employed to facilitate the inclusion of exon 7 in SMN2 transcripts, thereby enhancing the production of SMN2 protein [
137]. However, the limited delivery of the currently used ASO in the rostral spinal cord and brain has reduced therapeutic efficacy. Nusinersen, a modified 2′-MOE PS ASO, has been recently approved by the FDA for the treatment of SMA. Intrathecal injection of Nusinersen can significantly improve motor function and increase the lifetime of SMA patients [
138,
139]. However, this procedure is invasive and is linked to unpleasant post-lumbar puncture adverse effects for the patients [
137]. Therefore, to address this, PPMO trials have been conducted. Intravenous administration of Pip6A-PMO in the Taiwanese severe SMA mouse model increased mean survival and SMN2 expression in the brain and spinal cord and improved neuromuscular junction morphology [
140].
8. DG9: A CPP for Enhancing the Delivery and Cellular Uptake of ASO and Proteins
Although CPPs hold promise in facilitating the transport of biologically active cargo across cell membranes, including the notorious blood–brain barrier and other challenging barriers within the body, they also pose a number of difficulties and issues that require careful study. The primary obstacle to completing clinical trials for PPMO-based medications right now is their toxicity and immunogenicity. Toxicity can be variable depending on several factors, including species, treatment duration, frequency of systemic administration, dosage, exons skipped, and the cationic nature of the peptide [
83].
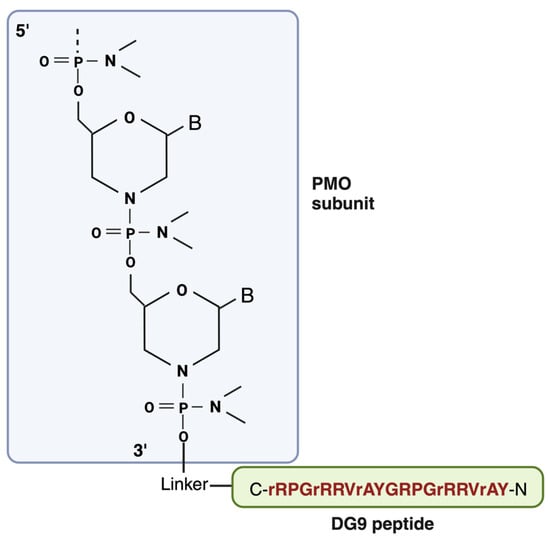
DG9 is a cell-penetrating peptide derived from the protein transduction domain (PTD) of the human Hph-1 transcription factor, which facilitates the cell membrane penetration of its protein cargos in the lungs (
Figure 4). Two of these Hph-1 domains constitute DG9 [
9]. In a study by Choi et al., it was shown that intraperitoneal injection of fusion proteins conjugated with the Hph-1 domain has enhanced delivery in a wide range of organs, including the heart and brain, which are apparently challenging to deliver. Additionally, according to the study, cell viability was not affected, and behavioral abnormalities, cytotoxic effects, and immunogenicity were not observed after 1.6 mg/kg of intravenous administration of Hph-1-fused protein into mice for 14 days or 100 μg of intraperitoneal injection two times a week for two weeks [
146]. The same author later reported another study where they used the same protein transduction domain (PTD) of the human Hph-1 transcription factor, but this time with two tandem sequences (HHph-1-PTD) and fused it with Foxp3 (the target protein of the study) protein to increase the cell permeability of Foxp3. In an in vitro study, HHph-1-Foxp3 was detected in the nucleus as well as in the cytoplasm within 30 min of transduction, suggesting that Foxp3 protein is efficiently delivered to cells and is localized in the nucleus. The delivery efficacy of HHph-1 was also proved in vivo, as HHph-1-Foxp3-treated mice lived longer and their phenotype improved compared to the control groups. They also found that two repeats of Hph-1-PTD (HHph-1) resulted in optimal intracellular transduction and rapid delivery compared to one Hph1 domain [
147].
The FDA has approved exon skipping as a promising therapy for DMD, which utilizes phosphorodiamidate morpholino oligomer (PMO) to target and modulate gene expression. Yokota’s research group has identified DG9 peptide conjugation as a powerful way of enhancing the exon-skipping efficacy of PMO in vivo [
9]. The positive aspect of the DG9 peptide used in this study is that it has a potentially better toxicity profile compared to other peptides. As mentioned earlier, peptide-conjugated PMOs have been found to induce dose-dependent toxic effects in preclinical studies, which are thought to be linked with their amino acid compositions [
83,
144,
150]. It has also been reported that substituting D-amino acid for L-amino acid in polymer-peptide conjugates attenuates anti-polymer antibody generation and toxicity and exhibits good tolerance in vivo even after repeated administration [
151]. Therefore, certain L-arginine residues in DG9 were converted to D-arginine (DG9 (sequence N-YArVRRrGPRGYArVRRrGPRr-C; uppercase: L-amino acids, lowercase: D-amino acids)) [
9]. This conversion has been shown to improve the viability of peptide-conjugated PMO-treated cells in vitro, along with increasing serum stability [
152].
9. Conclusions
Despite their huge potential, the poor biodistribution of nucleic acid-based medications has prevented them from being used clinically. As a result, cell-penetrating peptides have been thought of as a potential tactic to enhance passage across biological barriers and intracellular delivery, as covered in great detail in this chapter. However, there are a number of concerns that need to be resolved before CPPs are used in clinics, such as in vivo stability, immunogenicity, cellular toxicity, lack of selective intracellular uptake, and inability to escape from endosomes. Despite extensive research, the underlying mechanisms regulating the extravasation and cellular transport of these CPP-drug conjugates or complexes remain poorly understood. Undoubtedly, a deeper comprehension of these PPMOs’ limitations, pharmacodynamics, and mode of action will help create new CPP generations that are better able to target various tissues (and clinical diseases) and deliver their payload within the appropriate cellular compartment, giving patients hope for an improvement in their quality of life. The integration of DG9 into ASO-mediated therapy holds the potential to enhance cellular uptake and biodistribution of PMO, opening the door to more effective and precise treatments for a wide range of disorders. However, further research and development are necessary to fully realize the potential and long-term safety considerations. While DG9 has demonstrated proficiency in facilitating ASO transport into the cytoplasm, the precise underlying mechanism remains a topic requiring deeper exploration. This quest for mechanistic understanding holds promise as a compelling avenue for future investigative pursuits. With continued scientific inquiry, answers may emerge, potentially solidifying CPP-conjugated ASO-mediated therapy as a valuable asset within the arsenal of gene therapy strategies.
This entry is adapted from the peer-reviewed paper 10.3390/cells12192395