Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Dentistry, Oral Surgery & Medicine
Arecoline is the primary active carcinogen found in areca nut and has been implicated in the pathogenesis of oral squamous cell carcinoma (OSCC) and oral submucous fibrosis (OSF).
- arecoline
- nicotine
- acetylcholine receptors
- oral submucous fibrosis
- oral cancer
- mouth neoplasms
- oral squamous cell carcinoma
- areca nut
1. Introduction
Arecoline, an active carcinogenic alkaloid found in areca nut, has been recognized as an important factor in the pathogenesis of premalignant and malignant oral disorders [1], specifically, oral submucous fibrosis (OSF) and oral squamous cell carcinoma (OSCC). However, the precise molecular mechanisms underlying arecoline-induced OSF and OSCC development remain unclear [2,3].
OSF is characterized by progressive fibrosis and inflammation of the submucosal tissues [4]. It is a potentially malignant disorder that is associated with an increased risk of OSCC, a malignant neoplasm originating from the stratified squamous epithelium of the oral mucosa [5]. OSF progression to OSCC takes place in approximately 7–14% of patients [6]. OSCC is one of the most frequently reported malignancies in the world, especially in Taiwan and India, accounting for approximately 90% of all oral cavity cancers [7].
2. Role of Arecoline in Oral Carcinogenesis
Evidence of Increased Cell Growth and Proliferation
In vivo studies on murine models exposed to arecoline revealed the presence of squamous cell hyperplasia in the excised samples [8,18,19,20]. The introduction of arecoline-stimulated OSCC cell lines resulted in tumor growth [21]. Furthermore, certain studies explored the gene expressions associated with cellular proliferation and noted that arecoline promoted the gene expression of notch receptor 1 (NOTCH1), protein tyrosine kinase 6 (PTK6), and discoidin domain receptor tyrosine kinase 1 (DDR1) [8,10,12,22] while downregulating the tumor suppressor gene, retinoic acid receptor beta (RARB) [23].
Numerous in vitro studies have demonstrated that arecoline exposure leads to increased cell proliferation in various oral cell lines, including OSCC cells, oral keratinocytes, and gingival fibroblasts [8,10,24,25,26,27]. The levels of numerous biomarkers of cell proliferation, such as proliferating cell nuclear antigen (PCNA) and antigen kiel 67 (Ki67), were found to be elevated with arecoline treatment [8,24]. Studies that investigated gene expression found that arecoline triggers the upregulation of a wide range of genes and signaling pathways, including NOTCH1, MYC proto-oncogene (MYC), peroxiredoxin 2 (PRDX2), Wnt pathway (WNT), cysteine-rich angiogenic inducer 61 (CYR61), epidermal growth factor receptor/phosphoinositide 3-kinases (EGFR/Pl3k), and discoidin domain receptor 1 (DDR1) [8,12,22,28,29,30,31]. In contrast, the expression of tumor suppressor genes such as alcohol dehydrogenase, iron-containing 1 (ADHFE1), aldehyde dehydrogenase 1 family member A2 (ALDH1A2), dual specificity phosphatase 4 (DUSP4), and tumor protein p53 (TP53) was found to be downregulated upon exposure to arecoline [32,33,34].
Apoptosis/Cell Cycle Arrest
Caspase 8 (CASP8) was upregulated in mice models challenged with arecoline N-oxide (ANO) [20]. In vitro, arecoline induction was found to result in keratinocyte cell proliferation and inhibit apoptosis via PRDX2 gene overexpression [29], the elevation of CASP8 protein [20], the activation of mitogen-activated protein kinase 1/extracellular-signal-regulated kinase pathway (MEK1/ERK pathway) [36], and through triggering the ATM-dependent pathway, inducing arrest at mitosis [37,38]. There were observed increases in transcription factor Jun (c-jun) mRNA levels with fos proto-oncogene (c-fos) pathway activation, affecting cell cycle progression [36], but it was reported elsewhere that this did not induce c-fos mRNA expression [9].
Exposure of epithelial cells to arecoline was found to suppress viability and promote apoptosis and atrophy in a dose-dependent manner [26,39]. Arecoline inhibits epithelial cell proliferation and affects cell morphology, including cell cycle arrest in the G1/S phase and survival in a dose-dependent manner [35,40,41].
For SAS cancer cells, arecoline leads to cell death, apoptosis, and cell cycle arrest by stimulating checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2) phosphorylation [42].
In fibroblasts, arecoline inhibited the expression and function of tumor protein p53 (p53) and its downstream molecules [27,34,38], as well as down-regulated cyclin-dependent kinase inhibitor 1 (p21) and cyclin-dependent kinase inhibitor 1B (p27) [43], increased carbonic anhydrase IX (CAIX) expression [44], and led to cell-cycle exit [45]. Arecoline was also found to inhibit the tissue inhibitor of metallopeptidase 1 (TIMP-1) and tissue inhibitor of metalloproteinase 2 (TIMP-2) in fibroblasts in one in vitro study [42]; however, TIMP-1 was elevated in two other studies [46,47].
Promoting Invasion (Migration/Epithelial-to-Mesenchymal Transition (EMT)/Adhesion/Invasion)
Growth and invasion were promoted through the arecoline-induced upregulation of the NOTCH1 gene in mouse models [8,12]. EMT was promoted, activating invasion, and resulting in elevated PTK6 expression with E-cadherin (E-cad) suppression [10], which is deleted in esophageal cancer 1 (DEC1) upregulation, leading to focal adhesion kinase/serine/threonine kinase (FAK/AKT) [49], and through the upregulation of keratin 17 (Krt17) [52] in murine models.
In in vitro experiments, arecoline has been found to promote EMT in oral epithelial cells through DEC1 upregulation activating FAK/AKT downstream [49], the upregulation of proteasome activator complex subunit 3 (PA28γ), and the phosphorylation of MEK-1 [53] and was found to promote the expression of EMT-related genes [27,34]. Arecoline resulted in a dose- and time-dependent increase in zinc finger protein 1 (SNAI1) expression in human oral keratinocytes (HOKs) and OECM-1 [54]. The long-term exposure of buccal mucosal fibroblasts (BMFs) resulted in the dose-dependent upregulation of transcription factor zinc-finger E box-binding homeobox 1 (ZEB1) and the upregulation of insulin-like growth factor receptor 1 (IGF-R1) [55,56].
Fibrotic Alteration/Impaired Wound Healing
Arecoline challenge resulted in squamous cell hyperplasia, increased collagen deposition and fibrotic alteration, and also increased cervical lymph node (LN) metastasis in mice [8,18,19,48,49].
Arecoline-treated BMFs resulted in a dose-dependent increase in SLUG protein, leading to the increased expression of type I collagen [11]. Collagen production was increased through heat shock protein (HSP) 47 upregulation and altered matrix metallopeptidase 1, 2, and 9 (MMP-1, MMP-2, and MMP-9) expression [42,45,47,63,64]. Extracellular matrix (ECM) synthesis and secretion was increased through the upregulation of S100A4 gene expression [61]. Plasminogen activator inhibitor-1 (PAI-1) expression was dose-dependently elevated in arecoline-treated BMFs [65] but the increased expression of α-SMA (alpha smooth muscle actin) [52] was observed in one study, while decreased expression was seen in another [66].
Immune Responses and ROS/Antioxidant Activity
One in vivo study conducted on murine models observed increased antioxidant activity in heat shock protein 27 (HSP27) when the mice were exposed to arecoline [50].
Three in vivo studies investigated the effects of arecoline on different cytokines. Elevated levels of transforming growth factor-β (TGF-β) and interleukins (IL-1α, IL-1β, IL-10, and IL-17) were observed [12,21,48]. Tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ) were reduced in one of these studies [21].
Six in vitro studies observed ROS production in cells challenged with arecoline or its metabolites, including ANO, arecaidine, and arecoline N-oxide mercapturic acid (NOM) [12,26,54,68,69]. Antioxidant activities were shown to be reduced in some studies [12,69,70,71] but were increased in other studies [72,73,74].
Multiple in vitro studies have indicated the elevated expression of several inflammatory cytokines, including TGF-β, interleukins (IL-1α, IL-1β, IL-17α), serum amyloid A1 (SAA1), prostaglandin E2 (PGE2), and TNF-α [8,12,19,45,48,57,75,76,77,78]. However, one study observed a reduction in IL-6 and minimal changes in TNF-α [77]. One study mentioned the upregulation of programmed death-ligand 1 (PD-L1) in OSCC cells that were exposed to arecoline [28].
Genotoxicity and Epigenetics
The induction of DNA damage and the alteration of repair mechanisms are widely regarded as the central mechanisms responsible for arecoline-induced carcinogenesis. In vitro, arecoline stimulated an increase in O6-methyl-guanine-DNA methyltransferase (MGMT) expression in HOKs and an increase in the phosphorylation of H2AX variant histone (γH2AX) [19,24,45], as well as the induced markers of irreparable DNA double-stranded breaks in normal human oral fibroblasts and p53-binding protein 1 (53BP1) [45]. Low doses of arecoline induced elevated cell proliferation and DNA repair [24]; however, long-term and high-dose exposure reduced DNA repair [69]. Arecoline also resulted in the reduced expression of sirtuin 1 (SIRT1) mRNA [79].
In terms of epigenetic regulation, arecoline upregulated microRNA (miRNA) miR-211 expression in OSCC cell cultures [51]. miR-211-promoted OSCC was shown to repress gene transcription factor 12 (TCF12) and peroxiredoxin-like 2A (FAM213A) [51]. Arecoline exposure to cultured cells such as HOKs and OSCC cell lines led to a reduction in miR-1455 [13], miR30a, miR379, miR-203, miR-22, miR-200b, miR329, and miR410 and an increase in miR-23a, miR-886-3p, and miR-10b [13,27,31,32,42,76,79,81,82,83]. miR-23a overexpression was found to be associated with reduced double-stranded break repair [82], while miR-200 was shown to be involved in arecoline-related myofibroblast activities in BMFs and fBMFs [42].
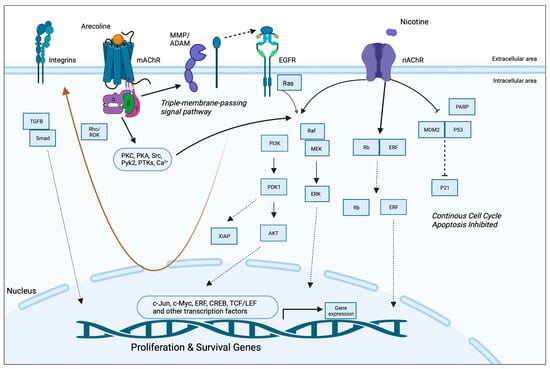
Arecoline-Mediated Acetylcholine Receptor Signaling in Oral Carcinogenesis
A study by Gareth Thomas’ group demonstrated that arecoline upregulated keratinocyte αvβ6 expression, a process modulated through the M(4) muscarinic acetylcholine receptor [57]. Arecoline-dependent αvβ6 upregulation promoted keratinocyte migration and induced invasion, raising the possibility that this mechanism may support malignant transformation. In another study, long-term nicotine-derived nitrosamine ketone (NNK) and arecoline exposure resulted in an increase in cancer stem cell properties, anti-apoptotic pathways, and a resistance to cisplatin in head and neck squamous cell carcinoma (HNSCC) cells in vitro [84]. The EGFR protein was pivotal in inducing tumor promotion and in impeding apoptosis in cancer cells by inducing phosphorylated AKT serine/threonine kinase 1 (pAKT) and nuclear factor kappa B (NFκB). While the authors pointed out that both NNK and arecoline exert agonist activity with the alpha-7-nicotinic acetylcholine receptor (α7-nAChR), the study did not directly investigate the role of nAChR in mediating the effects reported and, hence, was not included in the qualitative synthesis. Both studies, however, point to the possibility that arecoline promotes carcinogenesis via receptor-mediated mechanisms, an aspect that has not been captured in the available literature. The putative signaling pathways are depicted in Figure 2.
Figure 2. Putative pathways involving the receptor-mediated signaling of arecoline. Arecoline binds preferentially to muscarinic acetylcholine receptors (mAChR) but can also serve as a partial agonist of nicotinic receptors (nAChR). Two key molecular pathways involve EGFR and integrins. mAChR activates EGFR signaling via a so-called “triple-membrane-passing” pathway, whereby metalloproteinases cleave and activate EGF-like ligands, which, in turn, bind to EGFR and trigger downstream kinase signaling, including the Ras/Raf/MEK/ERK pathway. MAPK signaling can also be activated via canonical second messenger-mediated signals, as well as via nAChR. The two receptors also work synergistically to promote survival and inhibit apoptosis via PI3K/Akt and p21, respectively. Together, these pathways promote the expression of proliferation and survival genes, as well as migration/invasion and fibrosis/senescence via integrins and TGF-beta signaling, respectively (brown arrow). See Abbreviations part for the abbreviations and acronyms.
The Effects of Arecoline in the Oral Mucosa Could Be Mediated by the Local Cholinergic Axis
Previous research has convincingly demonstrated that both arecoline and guvacoline activate muscarinic acetylcholine receptors 1 and 3 (M1 and M3 mAChRs) [85], while only arecoline produces significant activation of the α4 nicotinic receptor and acts as a silent agonist of α7 nAChR [86]. A molecular docking simulation and antagonist co-exposure experiments also showed that arecoline has a strong affinity to muscarinic receptors M1–M4 [87]. Hence, it is likely that arecoline elicits cholinergic signals in the oral mucosa via the M2, M3, and M4 mAChR subtypes that are expressed in oral keratinocytes [88].
This keratinocyte cholinergic system has been shown to play a role in oral mucosal diseases [91] and also mediates nicotine toxicity in oral keratinocytes and in epithelial cancers [92]. It is now known that the nAChRs expressed on the cell membrane and mitochondria mediate both growth-promoting and anti-apoptotic effects synergistically. Other mechanisms associated with nicotine toxicity include the genotoxic action of reactive oxygen species [93]. With regard to mAChRs, accumulating evidence suggests that mAChR-dependent signaling pathways can promote cell proliferation and cancer progression [94].
In summary, there is increasing evidence that the non-neuronal cholinergic system in epithelial tissues is involved in carcinogenesis. Similar to the effects of nicotine, it is reasonable to speculate that AChR ligands, such as arecoline and other areca alkaloids, induce pro-tumorigenic effects in the oral mucosa via receptor-mediated signaling.
This entry is adapted from the peer-reviewed paper 10.3390/ph16121684
This entry is offline, you can click here to edit this entry!