Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Major depressive disorder (MDD) is a multifactorial psychiatric disorder characterized by at least one depressive episode for a minimum of 2 weeks. The gut is the biggest digestive, immune and endocrine organ of the human body and can sometimes be referred to as the second brain. The gut is a microbial organ, and it is estimated that our gut harbors about 1014 microorganisms.
- gut microbiota
- major depressive disorder
- physical exercise
1. Introduction
Major depressive disorder (MDD) is a multifactorial psychiatric disorder characterized by at least one depressive episode for a minimum of 2 weeks. The essential feature of a depressive episode is the presence of either a depressed mood or anhedonia accompanied by other neurocognitive and neurovegetative symptoms, such as diminished ability to think or concentrate and sleep disturbances [1]. Approximately 4–5% of the world population is affected by MDD [1]. Individuals with MDD are at a higher risk of suicidality [2], developing physical comorbidities (e.g., cardiovascular, stroke, diabetes, obesity) [3], and negative outcomes in other social aspects of life, such as education, employment and personal relationships [4].
MDD is a complex disorder whose mechanisms are not completely established [1]. Animal models have been an important strategy for investigating the molecular pathways implicated in the pathophysiology of MDD. These pathways include, but are not limited to, genetic and epigenetic alterations, neuroendocrine and immune disturbances, neuroplasticity impairments, mainly with regard to neurogenesis, and alterations in the microbiota–gut–brain axis [5]. An increased understanding of these biological processes may assist in developing new strategies for managing MDD.
The response to stressful stimuli, either psychological or physical, is mediated by the hypothalamic–pituitary–adrenal (HPA) axis [6]. The termination of the stress response is based on a negative feedback mechanism and is mediated by the connection between the hippocampus and the paraventricular nucleus of the hypothalamus [7]. A healthy response to stress requires both a rapid and vigorous response followed by the termination of such response [7]. However, a dysfunction of the HPA axis is present in individuals with MDD [1]. In addition, HPA axis activation may alter the composition of the gut microbiota and increase gastrointestinal permeability [8].
Individuals with MDD present higher serum levels of inflammatory markers [9]. Administration of lipopolysaccharide (LPS) in rodents has been used to mimic the inflammatory response and depressive symptoms observed in individuals with MDD [10]. LPS inflammatory effects extend from the periphery to the brain. Peripherally, pro-inflammatory cytokine production increases, and cytokines can subsequently reach the brain either via macrophage-like cell-mediated signaling or by crossing the blood–brain barrier (BBB), resulting in microglial activation [10]. Microglia are responsible for scavenging damaged brain tissue and pathogenic agents. At rest, they present ramifications that help their function as sentinels. In response to threats, microglia switch to their active phenotype. This results in the activation of the NOD-like receptor pyrin domain-containing-3 (NLRP3) inflammasome and the consequent release of the inflammatory cytokines interleukin (IL)-1β and IL-18, which contributes to neuroinflammation as shown in Figure 1 [11]. Accordingly, image studies have identified increased neuroinflammation in the brain of MDD patients as compared with controls [12,13].
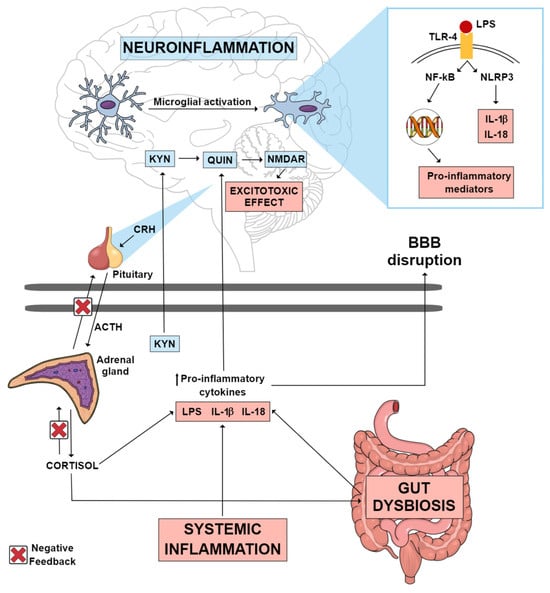
Figure 1. Possible inflammatory mechanisms thought to mediate neuroinflammation in MDD. Systemic inflammation, gut dysbiosis and an imbalance in the HPA axis can lead to an increase in circulating pro-inflammatory cytokines, which consequently may lead to the disruption of the blood–brain barrier. These pro-inflammatory cytokines can reach the CNS, where they can activate various mechanisms that culminate in neuroinflammation. Gut dysbiosis may be one of the factors contributing to the increase in circulating LPS. This endotoxin can reach the brain, where it may activate TLR-4 microglial receptors, promoting the transcription of pro-inflammatory mediators, as well as the activation of the NLRP3 inflammasome, which culminates in the production of IL-1 β and IL-18. A pro-inflammatory state in the CNS causes the KYN pathway to be directed towards the production of QUIN, an excitotoxic NMDAR agonist metabolite, instead of the production of KYNA, a neuroprotective NMDAR antagonist metabolite. Abbreviations: ACTH: adrenocorticotropic hormone; BBB: blood–brain barrier; CNS: central nervous system; CRH: corticotrophin-releasing hormone; HPA: hypothalamic–pituitary–adrenal; IL-18: interleukin 18; IL-1β: interleukin 1 beta; KYN: kynurenine; KYNA: kynurenic acid; LPS: lipopolysaccharide; MDD: major depressive disorder; NF-kB: nuclear factor kappa B; NLRP3: NOD-like receptor pyrin domain-containing-3; NMDAR: N-methyl-D-aspartate receptor; QUIN: quinolinic acid; TLR-4: toll-like receptor 4.
Another key inflammatory mechanism implicated in LPS-induced depressive-like behavior is the tryptophan (Trp)–kynurenine metabolism [14]. Trp metabolism can follow two directions: production of serotonin by tryptophan hydroxylase or production of kynurenine (KYN) by indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO) enzymes [15]. The conversion of Trp into KYN is activated under pro-inflammatory conditions [14]. KYN metabolites can be grouped into two pathways according to their effects: the excitotoxic pathway and the neuroprotective pathway [11]. KYN excitotoxic metabolites are induced in inflammatory states and include 3-hydroxy-kynurenine (3-HK), 3-hydroxy-anthralinic acid, quinolinic acid (QUIN) (a glutamate receptor agonist) and nicotinamide adenine dinucleotide (NAD+). Neuroprotective metabolites include kynurenic acid (KYNA), with opposing effects to QUIN by its antagonistic action on the N-methyl-D-aspartate (NMDA) glutamatergic receptor and reduction in extracellular glutamate release [11]. In MDD, there is an imbalance in the Trp-KYN pathway, favoring the production of neurotoxic over neuroprotective metabolites as illustrated in Figure 1 [16,17].
2. Gut Microbiota and MDD
The gut is the biggest digestive, immune and endocrine organ of the human body and can sometimes be referred to as the second brain [18]. The gut is a microbial organ, and it is estimated that our gut harbors about 1014 microorganisms [19]. The first signature microorganisms are acquired at birth and develop over the course of the first few years of life. Although it is easily modulated by host genetic and environmental factors, such as diet, stress and exposure to other microorganisms or antibiotics, the microbiota adapts to the host, carrying out several important metabolic and biochemical processes. Thus, gut microorganisms can directly influence human health [20].
The bidirectional connections between gut microorganisms and the brain through various biological systems are called the microbiota–gut–brain axis. These connections are fundamental for the maintenance of gastrointestinal, central nervous system (CNS) and microbial homeostasis and occur through direct and indirect communication via the autonomic nervous system, enteric nervous system, neuroendocrine system and immune system and signaling through microbial-derived metabolites and products, chemical transmitters, and neuronal pathways [21].
This close communication between the intestinal microbiota and the CNS is sensitive to several factors, mainly environmental ones, including diet, antibiotic use, stress and infections [22]. Disturbances in the homeostasis of these systems have already been related to the pathophysiology of MDD [23].
Clinical evidence has shown that MDD individuals have an altered microbiota composition when compared to healthy controls, including an increased Bacteroidetes/Firmicutes ratio, which is considered a dysbiosis signature [24]. Of note, a meta-analysis study has demonstrated a decreased abundance of the bacterial families Veillonellaceae, Prevotellaceae and Sutterellaceae, as well as the genera Coprococcus, Faecalibacterium, Ruminococcus, Bifidobacterium and Escherichia, and an increased abundance of the family Actinomycetaceae and the genus Paraprevotella in MDD patients compared to controls [23]. In addition, preclinical studies have shown that probiotics could improve depression-like phenotypes. For example, Tian et al. [25] reported that ingestion of the Bifidobacterium longum subspecies infantis strain CCFM687 improved stress-induced depressive-like behavior, increased BDNF levels and the abundance of butyrate-producing bacteria and modulated the HPA axis in mice. Also, treatment with Akkermansia muciniphila improved chronic-stress-induced depressive-like behavior, modulated corticosterone, dopamine, and BDNF levels and regulated gut microbiota and metabolites in mice [26].
The most direct communication between the gut and brain is through the vagus nerve (cranial nerve X) [27]. This peripheral nervous system (PNS) nerve has an important role in sensory and parasympathetic regulation of gut physiology regulating motility, digestion and tonic secretion of gastric mucus via the neurotransmitter acetylcholine (ACh) [28,29,30]. Vagal neurons synapse with intestinal enteroendocrine cells (EECs), such as neuropod cells that can release neurotransmitters including glutamate and serotonin, glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). These transmit sensory stimuli from the gut to the brain in milliseconds [20,31]. Several studies have suggested that abnormal composition of the gut microbiota is related to depressive and anhedonia-like phenotypes, and this is, at least in part, mediated by vagus nerve communication [32,33]. In fact, intraperitoneal injection of LPS in rats induced a depression-like phenotype that was abolished by subdiaphragmatic vagotomy [34]. Also, depression-like phenotypes, altered microbiota composition, systemic inflammation and downregulation of synaptic proteins in the medial prefrontal cortex were shown to be dependent on the subdiaphragmatic vagus nerve in mice exposed to LPS [35].
The gut microbiota is essential to the digestion of food, absorption of nutrients and production of metabolites like short-chain fatty acids (SCFAs), lipids, vitamins, bile acids, branched-chain amino acids, Trp and indole derivatives [36,37]. SCFAs are one of the most well-characterized metabolites produced by gut microbiota. They are saturated fatty acids with a carbon chain ranging from one to six atoms in length produced through the fermentation of dietary fiber in the colon, with acetate (C2), propionate (C3) and butyrate (C4) being the most common SCFAs [38]. After being absorbed by the colonocytes, SCFAs serve as an energy source, through the production of ATP in the mitochondria, being substrates for the synthesis of cholesterol and fatty acids. Moreover, SCFAs may improve gut and brain health and maintain the integrity of the intestinal and blood–brain barriers by regulating tight junction proteins, affecting mucus production and influencing gastrointestinal motility and appetite through the regulation of neuronal activity and intestinal hormones, such as GLP-1 and PYY. Also, SCFAs can protect from inflammation by modulating pro-inflammatory cytokines and chemokines, inhibiting nuclear factor kappa B (NF-kB) as well as histone deacetylases (HDACs) [39] and regulating microglial homeostasis in the CNS [40,41]. The main mechanism by which SCFAs seem to exert their effects is by activating G-protein-coupled receptors (GPRs) that are expressed in several cell types, including immune cells, adipocytes, skeletal and heart muscle cells, intestinal cells and brain cells [42,43,44]. More specifically, acetate, propionate and butyrate bind to GPR43 and GPR41 (also known as free fatty acid receptor 2 (FFAR2) and FFAR3, respectively), with butyrate also binding to GPR109A (also known as hydroxycarboxylic acid receptor 2 (HCAR2)) [45]. Among other functions, GPR43 and GPR41 are involved in the production and secretion of GLP-1 and PYY and exert anti-inflammatory and anti-tumorigenic activities [46,47].
Butyrate, the only known SCFA agonist for GPR109A, appears to be the principal agonist in the gastrointestinal tract [48], but this receptor can also be activated by niacin (also known as nicotinic acid and vitamin B3) and β-hydroxybutyrate [49,50]. Among other functions, GPR109A is involved in the reduction in chemokine and pro-inflammatory cytokine production, including IL-1β, IL-6 and tumor necrosis factor α (TNF-α) [51,52], as well as the inhibition of the NLRP3 inflammasome [53]. Furthermore, GPR109A can enhance the activity of adenosine monophosphate (AMP)-dependent kinase (AMPK) in microglia, resulting in the activation of sirtuin 1 (SIRT1), which inhibits NF-κB signaling via acetylation [54]. The nuclear factor (erythroid-derived) related factor-2 (Nrf2), which mediates antioxidant and anti-inflammatory signaling, has also been implicated in the pathways associated with the activation of this receptor [55]. Finally, GPR109A signaling has also been shown to be involved in the increase in several neurotrophic factors, including vascular endothelial growth factor (VEGF) and brain-derived neurotrophic factor (BDNF) [56,57]. All these pathways have been implicated in the pathophysiology of MDD, suggesting that butyrate production by the gut microbiota and activation of GPR109A may improve depressive symptoms.
SCFAs not only play an important role in CNS homeostasis by maintaining the integrity of the BBB but also have the ability to cross it, where they can regulate brain development, neuroplasticity, neurotransmitter synthesis, epigenetic factors and gene expression, as well as the immune response [21,40]. Indeed, sodium butyrate was shown to prevent microglia activation and depressive-like behaviors in mice [58], and in vitro studies have demonstrated its anti-inflammatory role through the decrease in LPS-induced microglial inflammation [59] in rat primary microglia cultures, hippocampal slices and co-cultures of rat cerebellar granule neurons, astrocytes, and microglial cells [60]. Furthermore, SCFAs have also been shown to reduce depressive-like behaviors by regulating HPA activity. In agreement, oral administration of acetate, propionate and butyrate was able to improve changes in intestinal permeability, HPA hyperactivity and anhedonia in mice submitted to repeated psychosocial stress [61].
As mentioned above, dysfunctions in neuroplasticity and neurogenesis have also been implicated in the pathophysiology of MDD. SCFAs have the capacity to modulate neurotrophins such as BDNF, nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF) that regulate the growth, survival and differentiation of neurons and synapses, thereby impacting neuroplasticity [62,63]. In fact, subcutaneous administration of sodium butyrate was shown to induce cell proliferation, migration and differentiation in the hippocampal dentate gyrus in a rat model of ischemia [64]. Moreover, a combination of sodium butyrate and pyridoxine promoted cell proliferation and neurogenesis in mice [65]. In addition, systemic administration of sodium butyrate has been reported to induce histone hyperacetylation in the hippocampus and frontal cortex, upregulate BDNF transcript levels and elicit antidepressant-like effects [66].
Another important metabolite of the gut microbiota is lactate, which is produced by the fermentation of dietary fibers by lactic acid bacteria, such as the genera Lactobacillus and Bifidobacterium. Lactate can be further converted into different SCFAs by several bacterial species, including Eubacterium hallii, Anaerostipes spp. and Veillonella spp. [67,68]. Studies have suggested that lactate can be absorbed and cross the BBB and has an important role in CNS, being used as an energy substrate by neurons and contributing to synaptic plasticity [69]. Several lines of evidence have indicated that lactate abnormalities may be associated with MDD. Indeed, an increased urine lactate concentration was observed in severe MDD patients [70]. Moreover, lactate was able to improve stress-induced depressive-like behavior, and this effect was associated with changes in the expression of target genes involved in serotonin receptor trafficking, astrocyte functions, neurogenesis, nitric oxide synthesis and cAMP signaling [71]. Further studies have also supported the antidepressant effect of lactate. Karnib et al. [72] demonstrated that lactate was able to improve depressive-like behaviors and mediate resilience to stress by modulating hippocampal levels and activity of HDACs in mice exposed to social defeat stress. In addition, Carrard et al. [73] showed that L-lactate administration reversed corticosterone-induced depressive-like behavior and promoted the proliferation and survival of new hippocampal neurons in adult mice. Furthermore, pharmacological inhibition of adult hippocampal neurogenesis abolished this effect, demonstrating the role of neurogenesis in the antidepressant-like effect of lactate. In fact, lactate is involved in regulating neuronal plasticity-related genes, including those coding for BDNF, activity-regulated cytoskeleton-associated protein (ARC) and VEGF [74,75].
The gut microbiota can also regulate Trp metabolism and the KYN pathway in MDD, promoting a decrease in KYN and an increase in QUIN levels [76]. In the brain, the enzyme kynurenine aminotransferase (KAT) that converts KYN into KYNA is localized mainly in astrocytes, and microglia and macrophages are responsible mainly for the production of QUIN by kynureninase under inflammatory conditions [77]. Some microbial components present within the gut microbiota, such as LPS and lipoteichoic acids, can activate toll-like receptors (TLRs) and have been identified as key factors in initiating Trp metabolism through the KYN pathway [78]. Furthermore, SCFAs can modulate intestinal barrier integrity and systemic inflammation, which are in turn associated with an altered KYN pathway [79]. In addition, butyrate plays a role in reducing intestinal IDO expression by inhibiting HDAC and IFN-gamma-dependent phosphorylation of signal transducer and activator of transcription 1 (STAT1) and, subsequently, the STAT1-driven transcriptional activity of IDO [80].
The modulation of the KYN pathway by the gut microbiota may be implicated in abnormal KYN levels found in MDD patients. In fact, fecal microbiota transplantation (FMT) from MDD patients to antibiotic-treated rats induced anhedonia and anxiety-like behaviors along with decreased gut microbiota richness and diversity and an elevated KYN/Trp ratio [81]. In addition, a reduction in the Firmicutes phylum and a reduction in SCFA synthesis, which has been associated with increased inflammation and the diversion of KYN metabolism to the neurotoxic pathway with the consequent production of QUIN, have been observed in MDD patients [82,83].
This entry is adapted from the peer-reviewed paper 10.3390/ijms242316870
This entry is offline, you can click here to edit this entry!