Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Huntington’s disease (HD), despite its extrinsic simplicity—only mutation in one gene underlies the pathogenesis—has no effective treatment today. Designing an anti-HD therapy that would eliminate etiology is a topical and relevant task of biomedicine.
- Huntington’s disease
- drug target
- PARP1 inhibitor
1. Introduction
The development of Huntington’s disease (HD) is associated with the expansion (multiplication) of trinucleotide CAG in the first exon of the huntingtin (HTT) gene [4]. The pathogenesis of the disease is based on the expression of the huntingtin protein having an abnormal conformation and of its derivatives, such as clusters of N-terminal fragments and protein aggregates [5,6]. The formation of protein bodies driven by various forms of the mutant huntingtin occurs in medium spiny neurons during the progression of the pathology and leads to their death. These phenotypic markers can be observed in postmortem brain samples from patients or animal models [7]. The disease is accompanied by behavioral symptoms, for instance, impaired motor functions and disturbances of mental and cognitive abilities [8].
What happens in HD at the molecular level? At the moment, aberrations are being revealed in such molecular pathways as the utilization of proteins and cellular structures (namely, the ubiquitin–proteasome system and autophagy), expression regulation of intracellular signaling, cytoskeleton-associated processes (e.g., cell transport and division), glutamatergic and dopaminergic synaptic signaling systems, and, finally, mitochondrial function; the latter dysfunction is combined with destabilization of energy homeostasis and oxidative stress, which leads to activation of pathways of a stress response and apoptosis [9,10].
2. Transcription Dysfunction
The most frequently, as shown by recent studies, transcriptional impairment affects neurotransmitter receptors, ion channels, and BDNF [11,12]. Aberrant huntingtin interacts with different transcription-regulating proteins (p53, CREB, CBP, and MSK-1, which control cell proliferation and DNA damage repair), PGC-1α, organelle and vesicle transporters, and dopamine 2 receptor–interacting proteins [13]. There is evidence that mHTT’s interactions with the transcription factors p53, CBP, CREB, SP1, NF-κB, REST, Foxp1, and HSF1 impede gene expression [14,15,16,17,18,19].
Normally, huntingtin binds to transcription factors that recognize a neuron-restrictive silencer element (NRSE) region of the genes necessary for life support of spiny neurons and thereby regulates their transport to the nucleus from the cytoplasm and their activity. Impairment of this interaction leads, in particular, to the shutdown of expression of the NRSE-containing genes BDNF and REST [20].
PGC-1α is involved in metabolic regulation and mitochondrial biogenesis. CREB-mediated transcription of PGC-1α is suppressed by mutant huntingtin. Conversely, recovery of the PGC-1α level impedes mHTT aggregation in striatal neurons [21,22]. mHTT inhibits SP1-dependent transcription as shown in postmortem brain samples from HD patients [23]. Additionally, mHTT mediates translocation of the REST protein to the nucleus and causes silencing of genes [24]. The interaction of mHTT with p53 leads to the expression of apoptotic genes [25].
Decreased activity of enzymes that promote chromatin remodeling has been demonstrated in HD. Phosphorylated CREB (a cAMP-dependent transcription factor) binds to a CRE region in the promoter of genes important for neuronal survival [26] and activates transcriptional coactivator CBP, thereby directing it to chromatin being remodeled to assemble a transcription factor complex. mHTT interacts with CBP and TAFII130, thus preventing their binding to the CRE region and interrupting the CRE-mediated transcription process [20,27].
In a recent paper, a set of genes differentially expressed in HD was presented [28]. As for human studies, investigators have found IGDCC3, a participant in nervous system development, and XKR4, a regulator of the apoptotic process during development. In postmortem brain samples, DNAJB1, HSPA1B, and HSPB1 are reported to be upregulated in all cell types except for medium spiny neurons. At the earliest stages of HD, there is a change in the expression of genes HSPH1 and SAT1. Differential expression analysis of genes of transcription regulators has revealed the upregulated factors TCF4, FOSL2, BCL6, ZBTB16, FOXP1, KLF15, RXRA, CUX1, CREBBP, and NFIA and the downregulated factors ZKSCAN1, MAX, E2F3, BCL11A, EGR1, FOXG1, TP53, HMG20A, JMJD1C, and STAT4 [28].
3. Systems of Clearance of Proteins and Other Cell Components
Heat shock proteins such as HSP70, HSP40, and HSP90 participate in the pathological processes of HD. It has been established in mouse models of HD that an increase in Hsp70 expression plays a neuroprotective role, and inhibition of Hsp90 results in mHTT degradation [29]. HSP40 and HSP70 suppress the formation of fibrillar aggregates from mutant forms of huntingtin, thus facilitating their refolding or, more often, their being directed to the ubiquitination and degradation system of the 26S proteasome complex [29,30,31,32]. Nevertheless, at the same time, heat shock proteins often lose their ability to perform their functions and get mixed up in growing mHTT aggregates. In this case, hydrolysis of the mutant protein by proteases occurs, which leads to the formation of N-terminal toxic fragments of mHTT [33,34]. These fragments exist in several forms: soluble monomers, oligomers, or aggregates. Therefore, mHTT generally disrupts the process of protein transfer by chaperones to proteasomes for both itself and other proteins.
4. Cytoskeleton Impairment: Intracellular Transport and Synaptic Transmission
The structural role of HTT is to combine microtubules with carrier proteins such as dynein and kinesin for transport pathways in the cell. The absence of this function in mHTT impairs intracellular trafficking, thereby having a strong effect on synaptic activity owing to a decrease in the transport of mitochondria, vesicles, and other structures along axonal microtubular pathways [35,36,37,38]. Thus, neurotransmission via receptors such as the GABAA (γ-aminobutyric acid receptor type A) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors deteriorates because mHTT impairs the ability of the HAP1 protein to mediate the binding of the KIF5 protein to the receptors implementing microtubule transport [39,40]. In addition, the transport of neurotrophic factor BDNF and its receptor TRKB, which ensure the survival of neurons, are disturbed too [41].
Transport failures, coupled with a decrease in energy, also diminish the reuptake of glutamate (a ligand of NMDA receptors), as a result of which excitotoxicity develops. It consists of hyperactivation of NMDA receptors and an enhancement of the flow of calcium ions into the cell; this alteration triggers the respiratory chain of electron transport. Excessive exposure to calcium induces upregulation of reactive oxygen species [42].
5. Mitochondrial Dysfunction
Mitochondria are a link between several pathological processes: disturbances of the electron transport chain, of metabolic processes, and of calcium homeostasis [11,12].
For ATP synthesis, transmembrane potential of mitochondria should not deviate from the optimal value. A decrease in the activity of respiratory chain complexes, especially complex II (succinate dehydrogenase) and complex III (ubiquinol-cytochrome c oxidoreductase), as well as impaired protein transport between mitochondria and cytosol, owing to the interaction of mHTT with TIM23, cause an aberration of membrane potential of mitochondria [43]. Furthermore, mHTT provokes mutations in mitochondrial DNA, which lead to heteroplasmy [44].
Problems also appear in metabolic pathways because it is reported that mHTT inhibits the binding of TAF4 to the PGC-1α gene promoter, which is involved in glucose metabolism and fatty acid β-oxidation [45]. The GAPDH protein binds to N-terminal regions of mHTT, thereby providing its enhanced delivery to the nucleus, and subsequently loses its activity after entering the mHTT aggregates [46].
Normally, electron leakage from the electron transport chain occurs mainly due to the functioning of complexes I and III resulting in the formation of reactive oxygen species, such as the superoxide anion and peroxides, which the antioxidant system can handle. In addition, the activities of enzymes in the Krebs cycle, for example, aconitase, are worsened [47].
Mutant HTT not only enhances the formation of reactive oxygen species but also disrupts the expression of proteins of one of the pathways of the antioxidant system: NRF2/ARE [48]. As mHTT interacts with CREB-binding proteins and TAF4, mHTT decreases the amount of coactivator PGC-1α, which negatively affects the expression of respiratory chain enzymes’ nuclear subunits, of the antioxidant system, of TFAM (a mitochondrial–DNA transcription regulator), of ATP synthase, and of superoxide dismutase [49].
By binding the MFN2 protein, N-terminal fragments of mHTT disturb the expression of genes necessary for maintaining mitochondrial morphology and mitogenesis in C. elegans models of HD. In addition, the whole mHTT protein accumulates in mitochondria and stimulates division of mitochondria by recruiting Drp1. It was recently shown that inhibition of the calcineurin–Drp1 pathway or upregulation of Opa1 promotes the survival of striatal neurons [50].
Mitochondria-associated membranes are microdomains enriched with inositol-1,4,5-triphosphate (IP3) receptors (IP3Rs), ryanodine receptors (RYRs), and molecular chaperones (such as glucose-regulated protein p75) that form functional complexes with voltage-gated anion-selective channels to ensure the transfer of Ca2+ from the endoplasmic reticulum to mitochondria [51].
mHTT depolarizes the mitochondrial membrane in several ways: it affects the outer and inner pores of mitochondria and induces the formation of ion channels, which changes the flow of protons through the inner membrane of mitochondria. Membrane depolarization, in turn, disrupts calcium buffering in mitochondria and promotes the opening of permeability pores (MPTP) at lower Ca2+ concentrations, thus leading to a cytochrome c release and mitochondrial swelling. Another way to increase the calcium concentration in the cytosol is the effect of mHTT on IP3 receptors [12,52].
6. Cell Death
Events taking place in mitochondria under the influence of N-terminal fragments of mHTT induce the release of cytochrome c, its binding to deoxy-ATP, APAF-1, and caspase 9, and formation of the apoptosome. This complex then activates caspases 3, 6, and 7, initiating caspase-dependent apoptosis [12].
Different caspases have dissimilar effects on the course of pathological processes: caspase 2 can cleave mHTT and generate toxic N-terminal fragments causing neurite degeneration; caspase 7 in a complex with mHTT activates other caspases, thereby stimulating apoptosis [53]; and caspase 8 cleaves off and activates the BCL2 (B-cell lymphoma 2) domain, whose activity induces caspase-independent apoptosis. mHTT also stimulates apoptosis in other ways: binding to the transcription factor p53 and increasing the expression of Bax and PUMA (p53-activated modulator of apoptosis) [54]. Additionally, the interaction of mHTT with HIP1 is weakened, which allows HIP1 to freely interact with proapoptotic proteins, for example, procaspase 8, which also provokes cell death.
Intensive production of reactive oxygen species caused by mitochondrial dysfunction results in DNA damage. Emergence of DNA double-strand breaks hyperactivates poly-ADP-ribose polymerase I (PARP1), which recruits DNA repair enzymes by synthesizing poly(ADP-ribose) chains [55,56]. Excessive accumulation of poly(ADP-ribose) triggers a process of cell death called “parthanatos”, during which proteins such as AIF and SMAC/DIABLO are released from mitochondria into the cytosol. AIF moves into the nucleus and causes DNA fragmentation and inhibition of poly(ADP-ribose) polymerase, thereby accelerating cell damage and destruction. SMAC/DIABLO binds to an X-linked apoptosis inhibitor protein and triggers apoptosis by inhibiting the antiapoptotic activity of the X-linked apoptosis inhibitor protein [56].
7. The Role of Astrocytes
Aggregates of mHTT are observed in astrocytes, although to a lesser extent than in spiny neurons [57]. Astrocytes are involved in the protection of neurons from excitotoxicity, in particular, by regulating the level of glutamate in the extracellular space through transporters such as EAAT2 and GLT1 [58,59]. In some cases, for example, with a certain number of glutamine repeats (polyQ), astrocytes exhibit signs characteristic of HD in neurons [60,61]. Inwardly rectifying K+ channels’ abundance is decreased in astrocytes, and therefore membrane potential diminishes as well [62]. Binding of mHTT to N-terminal myelin regulatory factor and inhibition of concurrent binding of CREB and TAF4 by mHTT reduce the expression of myelin core proteins and cause myelination deficiency and oligodendrocyte dysfunction [63].
8. Molecular Pathways of HD
Established pathways of HD can be found in databases of protein–protein interactions (PPIs) or molecular pathways and Gene Ontology. For instance, in the KEGG database (Kyoto Encyclopedia of Genes and Genomes, https://www.genome.jp/kegg/, accessed on 23 August 2023), there is information about the following pathways in this regard: Calcium signaling, Ubiquitin–proteasome system, Autophagy, Apoptosis, TNF signaling, Oxidative phosphorylation, Microtubule-based transport, and Transcription.
A comparison of these data with previously described pathways from recent literature reviews [12,52,64] indicates that there are several mechanisms in the pathogenesis of HD.
Mutations in the HTT gene lead to the loss of functions and to emergence of new types of activity of mHTT, the appearance of toxic N-terminal fragments during proteolytic activity, and mHTT aggregation. Collectively, these events disrupt the entire functional network of HTT and lead to failures in areas of natural functions of mHTT. In addition, due to the new functions of mHTT, N-terminal fragments, and the formation of physical obstacles in the form of protein aggregates, other cell processes fail too. In general, the following disturbed areas of cell physiology can be distinguished: cellular transport, systems of defense and homeostasis of the cell, and energy supply and metabolism (in particular, mitochondrial activities).
Cellular transport means the processes of physical motion of molecules or cellular structures, such as mitochondria, autophagosomes, or vesicles containing neurotransmitters. Structures of the transport system support both cell division (mitosis) and the mechanics of cell movement and migration, which involve the cytoskeleton and its associated proteins.
The second area of cell physiology that suffers because of mHTT is the systems of the cell response to stress, or defense systems, in particular, autophagy and various types of cell death (apoptosis and parthanatos) [65,66,67]. mHTT affects the defense systems not only at the cellular level but also in the whole body. Hyperactivation of activated microglia and cells of the peripheral immune system is induced by mHTT binding to transcription factors and leads to chronic inflammation and damage to neurons and peripheral tissues [68].
The consequences of impaired mitochondrial functions and their disturbed integrity are extensive. The aberrant activity of mHTT impedes mitochondrial biogenesis and results in a mitochondrial cytochrome c release, caspase activation, calcium dysregulation, and decreased energetic function [69]. Aside from a decrease in cell energy supply, (i) upregulation of reactive oxygen species, (ii) the induction of caspase-mediated apoptosis, and (iii) AIF-mediated parthanatos are among the key factors of neuronal death [66]. Based on the above facts, several basic pathways initiating mitochondrial dysfunction can be distinguished: the involvement of mHTT in the regulation of transcription, in transport of molecules into mitochondria, and in binding to enzymes and distortion of their functions. Pathological consequences of mHTT’s functions are summarized in Figure 1.
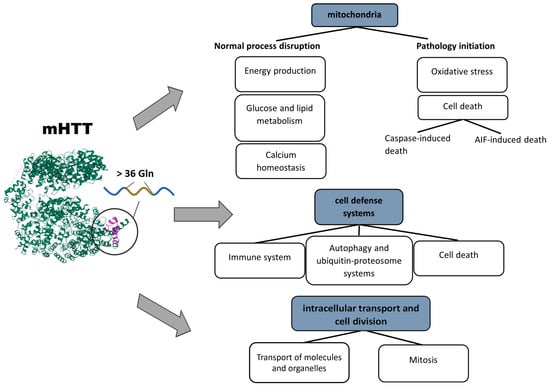
Figure 1. Pathological processes triggered by mHTT: mHTT causes a range of abnormalities in mitochondria, cell defense systems, intracellular transport, and cell division.
Some of the concerning questions are mHTT’s protein interactors and related processes. Addressing these questions should clarify not only the mechanism of HD but also new therapeutic targets. Above, we reviewed some impairments during HD development; however, the extent of mHTT involvement in these processes remains uncertain.
Recently, Wanker et al. reviewed mHTT’s PPIs and related pathways, such as axonal transport, autophagy, mTOR signaling, palmitoylation, mitochondrial fission, mitophagy, and transcription regulation [70]. Greco with the research group have analyzed PPIs of the HTT protein containing normal or expanded CAG repeats in a mouse HD model [71]. They have demonstrated that some processes associated with mHTT depend on the polyQ length and revealed the most common pathways of mHTT: processes related to vesicular trafficking and synaptic signaling. Accordingly, it was hypothesized that mHTT sequesters the key proteins that are necessary for the regulation of synapse morphology and neurotransmission.
Another research article on mHTT PPIs—with some severe limitations, e.g., in terms of polyQ length, experimental methods, and HTT species—has revealed the following PPI clusters associated with mutant huntingtin: protein modification, RNA splicing, mitochondria, granule membrane, macroautophagy, cytoplasmic vesicles, ion channel transport, and translation [72]. Each cluster involves the top KEGG pathways related to huntingtin.
9. HD Treatment
Today, most treatments reduce the motor and behavioral symptoms of HD; however, they do not address the underlying causes at the molecular level. In this section, we consider common therapeutic strategies targeting mHTT and pathological cell processes.
One of the strategies is to reduce the protein or mRNA level of mHTT. The latest therapeutic approaches are aimed at degradation or isolation of mRNA of mutant huntingtin, at suppression of its transcription, or at altering its processing [73,74]. Another way is to modify the mutant protein to decrease toxicity.
Antisense oligonucleotides (ASOs), by hybridizing with complementary mRNA, promote mRNA degradation and downregulation of both wild-type and mutant HTT. Testing on primates and rodents has yielded positive results [73], but such a therapy requires readministration for long-term suppression of mHTT synthesis. ASOs are also used for selective alteration of mRNA processing with the aim to prevent mHTT synthesis and cleavage and the formation of toxic N-terminal fragments [74]. To date, a number of ASO-based therapeutics have been developed and approved by the FDA: fomivirsen (Vitravene; for cytomegalovirus retinitis), mipomersen (Kynamro; for familial hypercholesterolemia), eteplirsen (Exondys 51; for Duchenne muscular dystrophy), and nusinersen (Spinraza; for spinal muscular atrophy).
Transcription repression is achieved also with the help of synthetic zinc finger protein (ZFP) repressors. These proteins consist of a zinc finger domain fused with some repressor, such as the KOX1 transcriptional repressor domain, and can selectively bind HTT containing expanded CAG repeats, thereby suppressing transcription. Testing of this therapy on the R6/2 model of HD has shown a reduced level of the mHTT protein and symptom stabilization [75]. It is necessary to conduct further trials to achieve greater selectivity and to check for side effects.
RNA interference machinery involving small interfering RNAs (siRNAs) [76], artificial microRNAs (miRNAs) [77], or short hairpin RNAs (shRNAs) [78] is also considered a potential HD therapy. Some siRNA- and miRNA-based therapeutics are in clinical trials, e.g., the AMT-130 trial (NCT04120493 and NCT05243017).
One more mRNA-targeting modality is small molecules mediating alternative splicing of mutant huntingtin mRNA: PTC518 (NCT 05358717, LMI070 (branaplam), NCT05111249).
The most direct way to decrease the mHTT level is to eliminate the mutant protein or to block its activity. Rapamycin, an inhibitor of the mTOR pathway, induces mHTT protein autophagy [79]. Cystamine and pridopidine are being tested to reduce protein aggregation. rAAV6-INT41 is an intrabody binding the polyP/proline-rich region of mHTT, thus reducing the aggregation in neurons of the R6/2 HD mouse model [80].
Another strategy of HD treatment is to target defective molecular pathways or functions. Generally, the following pathological processes are distinguished: impaired signal transduction, abnormal degradation of proteins, altered protein folding, transcriptional dysregulation, mitochondrial dysfunction [81], an axonal transport disturbance, and glial dysfunction [82].
Accordingly, based on currently known mechanisms of HD, numerous drugs are discussed in the literature. Firstly, a lot of studies are focused on synaptic dysfunction. To suppress excitotoxicity, drugs aimed at reducing glutamate receptors’ activity or availability of extracellular glutamate, e.g., Metamine [83] or BN82451 [84], have demonstrated efficacy in studies. Nonetheless, the safe concentration has not been determined yet. Another research field is aimed at regulating the dopamine level. FDA-approved drugs such as tetrabenazine [85] and valbenazine [86] inhibit vesicular monoamine transporter 2 (VMAT2), thereby reducing dopamine signaling. In addition, antibodies as a therapeutic modality are widely used: ANX005 (a monoclonal antibody) inactivates pathogenic complement cascade activation initiating neuroinflammation [87].
Mitochondrial dysfunction in HD has not been successfully corrected yet. Currently, resveratrol, which is an inhibitor of p53-mediated mitochondrial apoptotic oxidative stress [88], and PPARα agonist fenofibrate [89] are at the stage of clinical trials.
Most of described drugs in this field are intended to correct the molecular mechanisms whose malfunctions have been described. Anyway, a target can be identified among HTT’s or mHTT’s interactors directly: a loss of function or gain of function of mHTT may be an inducer of impairment. A low level of BDNF is associated with HTT dysfunction, leading to deficient synaptic transmission [90]. Histone hypoacetylation is one of the hallmarks of HD. mHTT can bind histone acetyltransferases, thus altering their functions. Sodium butyrate (a modulator of histone deacetylases) improves R6/2 mice’s motor symptoms and extends the lifespan [91].
Some researched drugs, such as neflamapimod [92] and minocycline [93], are utilized to ameliorate glial cells’ functions for preventing neuroinflammation.
Additionally, regardless of a target molecule, there is a type of therapy that solves the problem of degeneration through invasion. This method mainly concerns the maintenance of the population of medium spiny neurons. One of the approaches is to use stem cell-based therapy. Currently, researchers attempt to derive a pure population of a relevant type of cells not inducing an immune response: mesenchymal stem cells are at the clinical trial stage (NCT01834053) and induced pluripotent stem cell-derived neural stem cells have been tested on YAC128 HD mice [94].
Overall, the FDA has approved several drugs for HD. Among them, there are drugs designed to treat core symptoms, e.g., Austedo (deutetrabenazine), Xenazine (tetrabenazine), Valium, Risperdal (risperidone), and Ingrezza (valbenazine) (http://www.fda.gov/drugsatfda, accessed on 12 November 2023). Behavioral and cognitive abilities possess more complex mechanisms and regulations and have a variety of therapeutically targeted characteristics. Accordingly, to improve mental symptoms, drug repurposing is often used. FDA-approved drugs such as Clozaril (clozapine), Geodon (ziprasidone), Seroquel (quetiapine), Xanax (alprazolam), and Zyprexa (olanzapine) have originally been created for the treatment of schizophrenia or other mental disorders [95,96]. Some of these drugs have been approved for the treatment of HD, others are still at the clinical trial stage, but a search is still underway for a drug that could not only temporarily suppress symptoms or partially correct some molecular pathologies, such as aggregation, but has a long-term impact on multiple hallmarks of the disease with minimal adverse effects.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242316798
This entry is offline, you can click here to edit this entry!