1. Introduction
Colorectal cancer (CRC) is one of the most common malignancies, with about 1,881,000 new cases and 916,000 deaths from the disease worldwide every year [1], posing a serious threat to human health. Distant metastases are the leading cause of death due to treatment failure. The most likely sites for CRC to metastasize are the liver and lung [2], and the peritoneum is also a common site for CRC metastasis. Compared with the liver and lung, peritoneal metastasis (PM) in CRC cases usually means that patients lose the possibility of conversion therapy and have a worse prognosis. It has been reported that PM is discovered in approximately 13% of CRC cases, with 2% involving isolated PM and the remaining 11% being peritoneal and combined with other organs [3]. Patients who suffer from PM develop a higher proportion of BRAF mutations and worse survival. Due to the lack of effective treatment, the average survival of such patients was shorter than patients with liver-isolated metastasis (16.3 months (13.5–18.8) vs. 19.1 months (18.3–19.8)). Furthermore, PM combined with ≥2 sites of metastases has a 1.4-fold increased risk of death compared with PM alone [3].
The “seed and soil” idea states that tumor cells interact with milky spots, the peritoneal surface features, to create a milieu for tumor cell colonization and dissemination [4]. Based on the generation time of the primary lesion and PM, it can be divided into synchronous PM and metachronous PM. The former is defined as PM that is detected at the time of diagnosis of CRC or observed within 30 days after primary tumor resection; the latter is defined as PM observed 30 days after radical colorectal resection [5]. The spilling of cancer cells during tumor growth causes synchronous PM. Besides the intraperitoneal free cancer cells (IFCCs) caused by tumor itself, medical variables during surgery can shed cancer cells into the peritoneal cavity, causing metachronous PM.
2. Pathophysiological Process
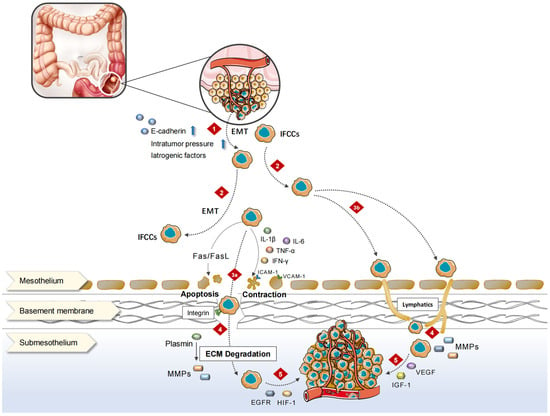
The peritoneum comprises the mesothelial cell layer, connective tissue, vascellum, and lymphatic tissue. CRC-PM can be achieved via hematologic, lymph node, or direct implantation. PM performs as a series of complex pathophysiological processes, the so-called “PM cascade”, involving multiple cellular, molecular, and genetic alterations, which create a special peritoneal microenvironment conducive to the CRC-PM.
The decrease in the expression or functional levels of intercellular adhesion molecule (ICAM), such as E-cadherin, causes CRC cells to disperse from the primary tumor into the peritoneal cavity, either singly or in clusters, and become IFCCs [
9]. Epithelial–mesenchymal transition (EMT) [
10] plays an important role in the acquisition of stem cell properties and motility/migration phenotypes by cancer cells. Cellular motility is augmented, extracellular matrix (ECM) and cell–cell adhesion are compromised, apic-base polarity is eliminated, and the cytoskeletal structure is reorganized throughout EMT. Under the contraction of actin contraction, each of these aggressive phenotypes promotes cancer cells to separate from the cancer nest, migrate, and invade [
11]. The loss of epithelial characteristics in aggressive frontier tumor cells and the adoption of mesenchymal-like phenotypes in CRC leads to enhanced aggressiveness [
12]. These modifications cause tumor cells to resemble normal cells during embryonic development, allowing tumor cells to adapt to shifting microenvironments and successfully spread.
The spread of IFCCs in the peritoneal cavity is not a random process. Under the influence of respiration, gravity, and intestinal peristalsis, intra-abdominal pressure changes and leads certain areas of the peritoneal cavity, such as the subdiaphragmatic region, the lesser sac, mesentery, diaphragm, and paracolic sulcus, to be at an increased risk of metastasis [
13]. On the one hand, IFCCs secrete various pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and interferon (IFN)-γ, which promote the high expression of ICAM, platelet endothelial cell adhesion molecule (PECAM), vascular adhesion molecule (VCAM) as well as other immunoglobulin superfamily cell adhesion molecules (IgCAMs) via human peritoneal mesothelial cells (HPMCs), and induce mesothelial cell contraction and roundness for the exposure of the basement membrane. On the other hand, the adhesion of IFCCs to HPMCs is mediated by the specific binding of CD43 (sialophorin) on the cell surface to the IgCAMs widely expressed by HPMCs above [
14]. Thereafter, HPMCs produce lysophosphatidic acid, which can further promote cancer cell adhesion and form “positive feedback”. In addition, IFCCs express CD44 molecules, a cell surface glycoprotein that interacts with hyaluronic acid secreted by mesothelial cells [
15], resulting in tumor cell mesothelial adhesion. A small number of tumor cells invade the mesothelium and induce HPMCs apoptosis through FasL/Fas [
16], disrupting peritoneal continuity and invading the subperitoneal space.
Matrix metalloproteinases (MMPs) play a significant role in CRC metastasis to the peritoneum. When tumor cells invade the subperitoneal space, surrounding cells like mesothelium, fibroblasts, inflammatory cells, and macrophages secrete MMPs, leading to the degradation of ECM [
17,
18]. In addition, the urokinase-type plasminogen activation system also disrupts the peritoneal barrier by activating pro-MMPs and degrading ECMs [
19]. Subsequently, tumor cells along with stromal cells generate various growth factors represented by epidermal growth factor receptor (EGFR) and insulin-like growth factor 1 (IGF-1) via autocrine and paracrine pathways to stimulate the continuous proliferation of cancer cells [
20]. Under a hypoxic environment, the expression of hypoxia-inducible factor 1α (HIF-1α) is significantly elevated in PM, further activating the transcription of angiogenesis-related genes including the vascular endothelial growth factor (VEGF) family [
21,
22], promoting the development of new microvessels and lymphatic vessels to obtain nutrients and oxygen, and finally leading to the formation of lymphatic metastasis and cancerous ascites (
Figure 1).
Figure 1. Mechanisms of CRC-PM. 1. In response to a variety of stimuli, proliferating cancer cells break from the original tumor and enter the peritoneal cavity. 2. IFCCs spread throughout the peritoneal cavity, finally implanting on the surface of distant HPMCs. 3. IFCCs cling to the peritoneum via mesothelial or lymphatic pathways. 3a. IFCCs release multiple kinds of pro-inflammatory cytokines that cause mesothelial cell contraction, rounding, or apoptotic shedding, exposing the basement membrane. HPMCs express a wide range of IgCAMs, which bind to glycoproteins on the surface of IFCCs and promote the adhesion of IFCCs to the peritoneum. 3b. IFCCs enter submesothelial lymphatic vessels through openings at the junction of two or more mesothelial cell-lymphatic pores, attaching to the peritoneum. 4. IFCCs invade the subperitoneal region and destroy the ECM via MMPs, causing the peritoneal barrier to rupture. 5. Neovascularization occurs in the tumor-promoting microenvironment created by different metastasis-associated variables, and cancer cells proliferate to form metastatic foci.
Tumor-associated fibroblasts (CAFs) are the most prevalent cell type in the tumor microenvironment (TME) and the hub of tumor mesenchyme cell communication. Lipid metabolism reprogramming also occurs in CAFs, whose secreted fatty acids and phospholipids are taken up by CRC cells and can promote CRC cell migration [
23]. CAFs can upregulate fatty acid oxidation rate-limiting enzyme CPT1A to actively oxidize fatty acids, thereby promoting CRC-PM [
24]. In addition, they can promote the migration of CRC-PM through the upregulation of unsaturated acyl chains in phosphatidylcholine to increase cell membrane fluidity, thereby increasing CRC cell migration and intraperitoneal spreading. Treatment with sodium palmitate (C16:0) reduced CAFs-induced changes in cell membrane fluidity and inhibited tumor growth and intraperitoneal spreading [
25], uncovering new opportunities for the future treatment of CRC-PM.
PM is caused by a multifactorial, multi-stage, and multi-gene interaction between tumor biology, peritoneal biology, and biological cytokines. Identifying and exploring the mechanism and theory underlying CRC-PM will help clinicians prevent and treat it better.
3. Molecular Biology Characterization
Although the molecular features of CRC-PM are not yet well understood enough to influence treatment decisions, a better understanding of molecular biology, including genomics, transcriptomics, and proteomics, can help quantify the risk of PM in individual patients after initial radical surgery and may be useful in predicting the benefits of specific drugs, advancing effective drug development and improving patient prognosis.
3.1. High-Frequency Mutations in PM
Precision medicine is based on individual bioinformatics using high-throughput technologies such as next-generation sequencing (NGS). In a genome-wide analysis by the Cancer Genome Atlas Network in 2012, mutations with high frequency in CRC primary foci included WNT (APC, CTNNB1, SOX9, TCF7L2, DKK, AXIN2, FBXW7, ARID1A, and FAM123B), PI3K, RAS-MAPK (IGF2, IRS2, PIK3R1, PIK3CA, PTEN, KRAS, NRAS, BRAF, ERBB2, and ERBB3), transforming growth factor-β (TGF-β) (TGFBR1, TGFBR2, ACVR2A, ACVR1B, Smad2, SMAD3, and Smad4), and p53 (53 and ATM) [
26]. Although CRC-PM differed in mutation frequency, the mutation pattern was similar to primary CRC: TP53 (median 54%, 33–75%), KRAS (45%, 20–58%), APC (44%, 31–57%), Smad4 (22%, 15–29%), BRAF (15%, 6–36%), and PIK3CA (13%, 9–14%) [
26].
There are some discrepancies in the current studies on KRAS and BRAF in CRC-PM. Some studies have suggested that in patients with CRC-PM, the rates of KRAS and BRAF mutations are similar to those of primary CRC [
26,
27,
28,
29,
30], but others have found that PM has a higher rate of BRAF mutations (25–36%) than primary CRC (5–10%) [
31,
32]. In some studies, BRAF mutations are a negative prognostic marker for patients with CRS/HIPEC [
28,
29,
33]. However, it has also been shown that BRAF mutation status does not influence survival [
34]. There are similar differences in KRAS. KRAS and KRAS codon 12 mutation was significantly associated with PM [
35]. RAS mutations also affected survival after CRS/HIPEC [
27,
36] and their mutation status could be used as a marker for predicting peritoneal recurrence [
37], while other studies showed the opposite [
28,
29,
34].
BRAF-mutant tumors in metastatic colorectal cancer (mCRC) have a greater incidence of PM and are substantially linked to microsatellite instability (MSI) [
38]. Compared to individuals with low-frequency MSI (MSI-L)/microsatellite stability (MSS) CRC, those with high-frequency MSI (MSI-H) CRC had a higher chance of developing PM [
39]. The expression of PD-1, tumor mutation load, and MSI-H were lower in CRC-PM than in primary tumors [
30], suggesting that CRC-PM has a unique molecular expression profile. Microsatellite (MS) status is also a stratification factor for CRC-PM patients undergoing CRS/HIPEC, with MSI having a better prognosis [
34,
40], which may be one of the reasons for the differences between KRAS and BRAF in different studies.
PIK3CA mutation was a protective factor against PM, and it was not only significantly associated with a lower chance of PM at the initial diagnosis of mCRC (OR 0.10, 95% CI 0.01–0.79) but it also dramatically reduced the risk of PM at any time thereafter (HR 0.31, 95% CI 0.11–0.86) [
41]. But, a recent study indicated that PI3K pathway modifications after CRS in CRC-PM patients were related to a drop in recurrence-free survival (RFS, 5 vs. 13 months), it represented a poor prognostic indicator, and it was a new molecular subtype of PM associated with early recurrence [
42].
In primary CRC, increased mutation burden in the tumor suppressor gene FBXW7 is associated with a lack of distant metastasis and increased expression of T cell proliferation and antigen presentation [
43]. In contrast, FBXW7 mutation burden is lower in PM [
30], which means it may play a role in the CRC-PM.
Among the genes repeatedly mutated by NGS (TP53, APC, Smad4, PIK3CA, and FBXW7), only the APC mutation (36.8%) was lower than the general level of mCRC, which negatively correlated with CRC-PM and suggested better survival. However, it is unclear whether this is a CRC-PM-specific alteration or the result of a higher proportion of right hemi-primary foci [
44]. The role of APC mutations in the occurrence and progression of CRC-PM needs to be further explored in additional studies.
CRC-PM showed significant molecular heterogeneity. In the same individual, key driver genes such as KRAS, APC, and TP53 mutations were homogeneous across samples from CRC-PM patients, special AT-rich sequence-binding protein 2 (SATB2) lacked expression in most cases, while less common mutations such as RNA-binding motif protein 3 (RBM3) showed significant heterogeneity in different samples from the same patients. Similarly, copy number variation (CNV) is heterogeneous both within and between patients [
45]. There are a few studies on CNV in CRC-PM; small sample studies have reported increased expression of 5P and 12P genes in PM compared to primary CRC foci or liver metastasis (LM) in the past [
46,
47].
Due to its varying molecular expression patterns from the initial lesion, more research is needed to determine the genome/epigenomic profile of CRC-PM, which will aid in patient prognosis and treatment.
3.2. Consensus Molecular Subtypes (CMS)
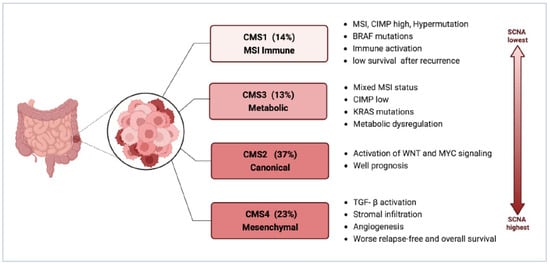
CMS4 might be the major phenotype of CRC-PM. CMS staging has a clear biological interpretation and is currently the most reliable classification system for CRC. It is also the basis for future clinical stratification and subtype-based targeted interventions, as shown in
Figure 2 [
48,
49].
Figure 2. Consensus molecular subtypes of CRC (Created with
biorender.com; accessed on 7 November 2023). Abbreviations: CIMP, CpG island methylator phenotype; MSI, microsatellite instability; SCNA, somatic copy number alterations; TGF-β, transforming growth factor-β.
Among the four types of CMS, CMS4 has the largest association with CRC-PM, which is clearly attributed to its molecular properties: enhanced tumor cell invasiveness due to TGF-β activation mediating EMT, as well as abundant and active angiogenesis, all of which constitute favorable conditions for PM. This has been validated in sequencing analyses based on primary CRC and PM tissue samples.
In the study by Ubink et al., CRC-PM was not only associated with poor histopathological features, such as a higher proportion of mesenchymal components in primary tumors and metastases, a low degree of differentiation in primary tumors, and high levels of tumor budding, but also a significantly higher percentage of CMS4 in primary tumors with PM (60% vs. 23%,
p = 0.002) and in PM, CMS4 enrichment was even higher (21/28, 75%), with at least one CMS4-positive tumor site was present in 15 out of the 16 patients with paired tumors [
31]. The higher proportion of mesenchymal, and stronger propensity for hematogenous metastasis makes such tumors appear to have some of the characteristics of sarcomas and thus may benefit from therapies targeting the tumor mesenchymal, such as anti-vascular therapy. It is important to note that there is considerable heterogeneity in tumor CMS4 status within patients, with inconsistent CMS4 classification between primary tumors and PM in 50% of patients [
31], suggesting that molecular phenotypes and signaling change markedly during CRC-PM and the CMS typing remains a limitation for the identification of molecular biological properties of CRC-PM.
It was also discovered in later research that nearly all tissue biological samples from patients with CRC-PM’s original tumors and PM foci were CMS4 categorized, but the expression of CMS4 recognition genes varied greatly between groups and was significantly higher in PM than in primary tumors, thereby distinguishing a population group with a poor prognosis. Moreover, the CMS4 status of CRC-PM and the high reductive capacity due to increased glutathione synthesis also led to oxaliplatin (OXL) resistance [
50]. In addition, this study demonstrated that 15 Hallmark pathways, including TGF-β signaling, angiogenesis, complement activation, and EMT, were expressed at significantly higher levels in PM than in their corresponding primary tumors, while WNT signaling and MYC target gene expression were significantly reduced. The presence of immature dendritic cells (DCs), monocytes, macrophages, and natural killer (NK) cells was significantly more characteristic in PM compared with their primary tumors [
48,
51]. These are all characteristics of CMS4 CRC and reflect the more extreme mesenchymal phenotype of CRC-PM. ZEB1 is a major regulator of EMT and can be used to identify CMS4 CRC in situ [
52]. So the highly mesenchymal phenotype of CRC-PM may be associated with their high expression of ZEB1.
However, there are different views. In Barriuso’s study, CMS2 was the subtype that comprised the highest proportion and had prognostic predictive value [
53]. The difference in the results shown may be attributed to the fact that both studies had small sample sizes and used assays built on different platforms.
More transcriptomic or proteomic studies should be encouraged to further clarify the complex downstream signaling pathways and the potential differences between CRC-PM and other metastatic types. This is necessary because the above findings are only a preliminary investigation of the molecular biology of CRC-PM, and most of the studies had small sample sizes. Furthermore, the samples collected were from a subset of patients with indications for surgery, which introduces selection bias. Many subjects also had distant metastases from other sites or included solid tumors of completely different pathological types, such as peritoneal pseudomyxoma and appendiceal malignancies. Additionally, the experiment’s selection of microarray and sequencing panels exhibited significant variability, with all these factors leading to divergent results among studies. The lack of an in-depth exploration of the underlying mechanisms behind the appearances further complicates the understanding of CRC-PM. To address these limitations, it is crucial to establish large, representative patient cohorts and standardized sample collection, processing, and analysis. Such efforts would not only improve the prognosis of these refractory patients but also guide individualized therapy.
This entry is adapted from the peer-reviewed paper 10.3390/cancers15235641