According to recent evidence, some groups of semaphorins (SEMAs) have been associated with cancer progression. These proteins are able to modulate the cellular signaling of particular receptor tyrosine kinases (RTKs) via the stimulation of SEMA-specific coreceptors, namely plexins (plexin-A, -B, -C, -D) and neuropilins (Np1, Np2), which share common domains with RTKs, leading to the coactivation of the latter receptors. MET, ERBB2, VEGFR2, PFGFR, and EGFR, among others, represent acknowledged targets of semaphorins that are often associated with tumor progression or poor prognosis. In particular, higher expression of SEMA6 family proteins in cancer cells and stromal cells of the cancer niche is often associated with enhanced tumor angiogenesis, metastasis, and resistance to anticancer therapy. Notably, high SEMA6 expression in malignant tumor cells such as melanoma, pleural mesothelioma, gastric cancer, lung adenocarcinoma, and glioblastoma may serve as a prognostic biomarker of tumor progression. To date, very few studies have focused on the mechanisms of transmembrane SEMA6-driven tumor progression and its underlying interplay with RTKs within the tumor microenvironment. The growing evidence is presented in the literature on the complex and shaping role of SEMA6 family proteins in cancer responsiveness to environmental stimuli.
- SEMA6
- plexins
- RTKs
- VEGF
- VEGFR1/2
- tumor
- dendritic cells
1. Semaphorin 6A
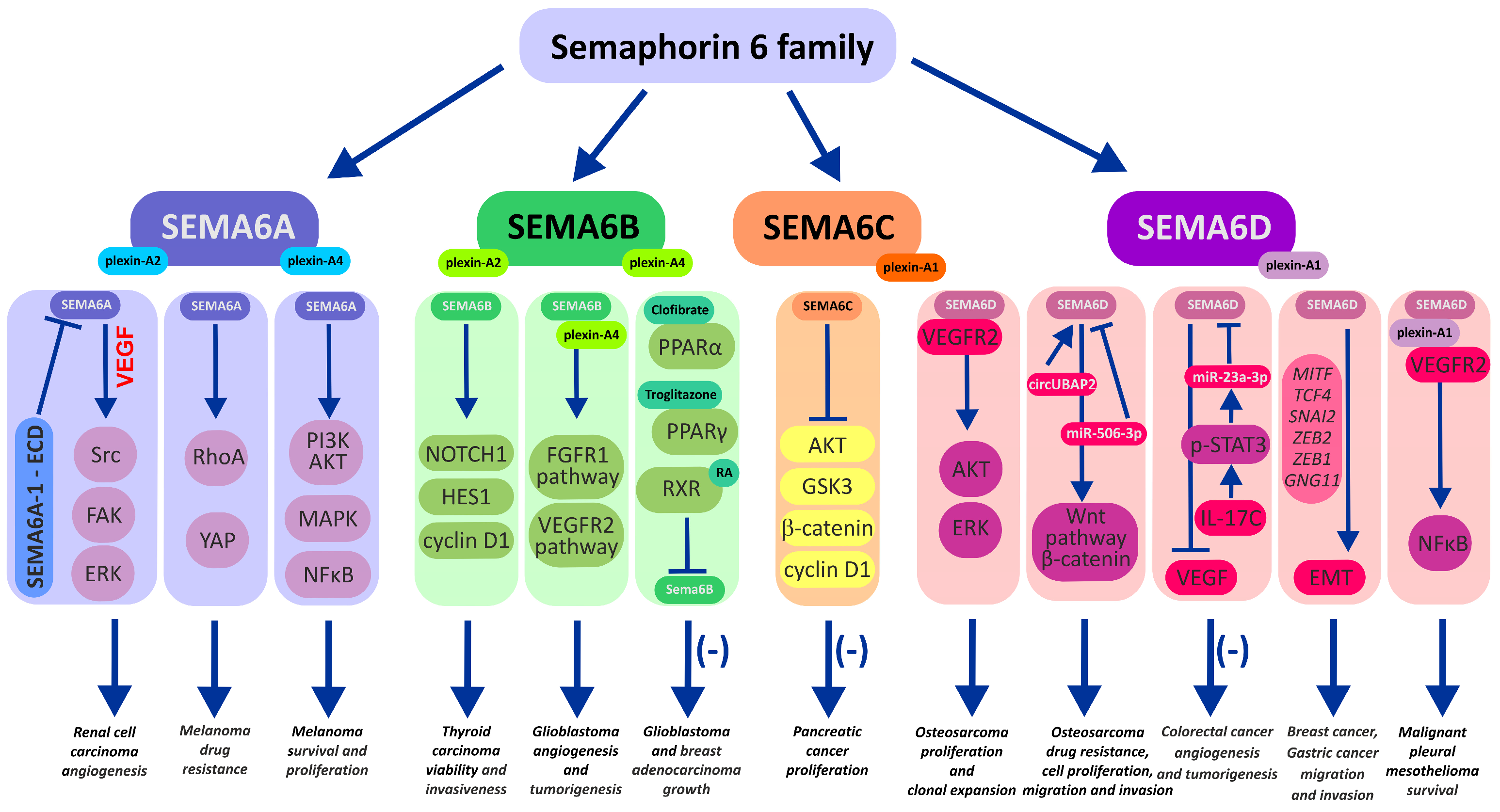
2. Semaphorin 6B
3. Semaphorin 6C
4. Semaphorin 6D in the Immunological Landscape of Tumors
This entry is adapted from the peer-reviewed paper 10.3390/cancers15235536
References
- Curley, J.L.; Catig, G.C.; Horn-Ranney, E.L.; Moore, M.J. Sensory axon guidance with semaphorin 6A and nerve growth factor in a biomimetic choice point model. Biofabrication 2014, 6, 035026.
- Xu, X.M.; Fisher, D.A.; Zhou, L.; White, F.A.; Ng, S.; Snider, W.D.; Luo, Y. The transmembrane protein semaphorin 6A repels embryonic sympathetic axons. J. Neurosci. 2000, 20, 2638–2648.
- Rünker, A.E.; Little, G.E.; Suto, F.; Fujisawa, H.; Mitchell, K.J. Semaphorin-6A controls guidance of corticospinal tract axons at multiple choice points. Neural Dev. 2008, 3, 34.
- Segarra, M.; Ohnuki, H.; Maric, D.; Salvucci, O.; Hou, X.; Kumar, A.; Li, X.; Tosato, G. Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood 2012, 120, 4104–4115.
- Dhanabal, M.; Wu, F.; Alvarez, E.; McQueeney, K.D.; Jeffers, M.; MacDougall, J.; Boldog, F.L.; Hackett, C.; Shenoy, S.; Khramtsov, N.; et al. Recombinant semaphorin 6A-1 ectodomain inhibits in vivo growth factor and tumor cell line-induced angiogenesis. Cancer Biol. Ther. 2005, 4, 659–668.
- Wolter, M.; Werner, T.; Malzkorn, B.; Reifenberger, G. Role of microRNAs Located on Chromosome Arm 10q in Malignant Gliomas. Brain Pathol. 2016, 26, 344–358.
- Tian, R.; Hu, J.; Ma, X.; Liang, L.; Guo, S. Immune-related gene signature predicts overall survival of gastric cancer patients with varying microsatellite instability status. Aging 2020, 13, 2418–2435.
- Lim, H.-S.; Kim, C.S.; Kim, J.-S.; Yu, S.-K.; Go, D.-S.; Lee, S.A.; Moon, S.M.; Chun, H.S.; Kim, S.G.; Kim, D.K. Suppression of Oral Carcinoma Oncogenic Activity by microRNA-203 via Down-regulation of SEMA6A. Anticancer Res. 2017, 37, 5425–5433.
- Yu, S.; Li, N.; Wang, J.; Fu, Y.; Huang, Y.; Yi, P.; Chen, R.; Tang, D.; Hu, X.; Fan, X. Correlation of Long Noncoding RNA SEMA6A-AS1 Expression with Clinical Outcome in HBV-Related Hepatocellular Carcinoma. Clin. Ther. 2020, 42, 439–447.
- Song, Z.-B.; Yu, Y.; Zhang, G.-P.; Li, S.-Q. Genomic Instability of Mutation-Derived Gene Prognostic Signatures for Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2021, 9, 728574.
- Liu, H.; Brannon, A.R.; Reddy, A.R.; Alexe, G.; Seiler, M.W.; Arreola, A.; Oza, J.H.; Yao, M.; Juan, D.; Liou, L.S.; et al. Identifying mRNA targets of microRNA dysregulated in cancer: With application to clear cell Renal Cell Carcinoma. BMC Syst. Biol. 2010, 4, 51.
- Loria, R.; Bon, G.; Perotti, V.; Gallo, E.; Bersani, I.; Baldassari, P.; Porru, M.; Leonetti, C.; Di Carlo, S.; Visca, P.; et al. Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget 2015, 6, 2779–2793.
- Lv, W.; Zhan, Y.; Tan, Y.; Wu, Y.; Chen, H. A combined aging and immune prognostic signature predict prognosis and responsiveness to immunotherapy in melanoma. Front. Pharmacol. 2022, 13, 943944.
- Loria, R.; Laquintana, V.; Scalera, S.; Fraioli, R.; Caprara, V.; Falcone, I.; Bazzichetto, C.; Di Martile, M.; Rosanò, L.; Del Bufalo, D.; et al. SEMA6A/RhoA/YAP axis mediates tumor-stroma interactions and prevents response to dual BRAF/MEK inhibition in BRAF-mutant melanoma. J. Exp. Clin. Cancer Res. 2022, 41, 148.
- Zhao, J.; Tang, H.; Zhao, H.; Che, W.; Zhang, L.; Liang, P. SEMA6A is a prognostic biomarker in glioblastoma. Tumour Biol. 2015, 36, 8333–8340.
- Chen, L.-H.; Liao, C.-Y.; Lai, L.-C.; Tsai, M.-H.; Chuang, E.Y. Semaphorin 6A Attenuates the Migration Capability of Lung Cancer Cells via the NRF2/HMOX1 Axis. Sci. Rep. 2019, 9, 13302.
- Shen, C.-Y.; Chang, Y.-C.; Chen, L.-H.; Lin, W.-C.; Lee, Y.-H.; Yeh, S.-T.; Chen, H.-K.; Fang, W.; Hsu, C.-P.; Lee, J.-M.; et al. The extracellular SEMA domain attenuates intracellular apoptotic signaling of semaphorin 6A in lung cancer cells. Oncogenesis 2018, 7, 95.
- Hasan, A.N.; Ahmad, M.W.; Madar, I.H.; Grace, B.L.; Hasan, T.N. An in silico analytical study of lung cancer and smokers datasets from gene expression omnibus (GEO) for prediction of differentially expressed genes. Bioinformation 2015, 11, 229–235.
- Lu, T.-P.; Tsai, M.-H.; Lee, J.-M.; Hsu, C.-P.; Chen, P.-C.; Lin, C.-W.; Shih, J.-Y.; Yang, P.-C.; Hsiao, C.K.; Lai, L.-C.; et al. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2590–2597.
- Prislei, S.; Mozzetti, S.; Filippetti, F.; De Donato, M.; Raspaglio, G.; Cicchillitti, L.; Scambia, G.; Ferlini, C. From plasma membrane to cytoskeleton: A novel function for semaphorin 6A. Mol. Cancer Ther. 2008, 7, 233–241.
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803.
- Andermatt, I.; Wilson, N.H.; Bergmann, T.; Mauti, O.; Gesemann, M.; Sockanathan, S.; Stoeckli, E.T. Semaphorin 6B acts as a receptor in post-crossing commissural axon guidance. Development 2014, 141, 3709–3720.
- Cordovado, A.; Schaettin, M.; Jeanne, M.; Panasenkava, V.; Denommé-Pichon, A.-S.; Keren, B.; Mignot, C.; Doco-Fenzy, M.; Rodan, L.; Ramsey, K.; et al. SEMA6B variants cause intellectual disability and alter dendritic spine density and axon guidance. Hum. Mol. Genet. 2022, 31, 3325–3340.
- Tawarayama, H.; Yoshida, Y.; Suto, F.; Mitchell, K.J.; Fujisawa, H. Roles of semaphorin-6B and plexin-A2 in lamina-restricted projection of hippocampal mossy fibers. J. Neurosci. 2010, 30, 7049–7060.
- Küry, P.; Abankwa, D.; Kruse, F.; Greiner-Petter, R.; Müller, H.W. Gene expression profiling reveals multiple novel intrinsic and extrinsic factors associated with axonal regeneration failure. Eur. J. Neurosci. 2004, 19, 32–42.
- Castellotti, B.; Canafoglia, L.; Freri, E.; Tappatà, M.; Messina, G.; Magri, S.; DiFrancesco, J.C.; Fanella, M.; Di Bonaventura, C.; Morano, A.; et al. Progressive myoclonus epilepsies due to SEMA6B mutations. New variants and appraisal of published phenotypes. Epilepsia Open 2023, 8, 645–650.
- Li, Q.; Liu, M.; Huang, D.-P.; Li, T.; Huang, J.; Jiang, P.; Ling, W.-H.; Chen, X.-Q. A De Novo SEMA6B Variant in a Chinese Patient with Progressive Myoclonic Epilepsy-11 and Review of the Literature. J. Mol. Neurosci. 2021, 71, 1944–1950.
- Xiaozhen, S.; Fan, Y.; Fang, Y.; Xiaoping, L.; Jia, J.; Wuhen, X.; Xiaojun, T.; Jun, S.; Yucai, C.; Hong, Z.; et al. Novel Truncating and Missense Variants in SEMA6B in Patients With Early-Onset Epilepsy. Front. Cell Dev. Biol. 2021, 9, 633819.
- Herzog, R.; Hellenbroich, Y.; Brüggemann, N.; Lohmann, K.; Grimmel, M.; Haack, T.B.; von Spiczak, S.; Münchau, A. Zonisamide-responsive myoclonus in SEMA6B-associated progressive myoclonic epilepsy. Ann. Clin. Transl. Neurol. 2021, 8, 1524–1527.
- Duan, J.; Chen, Y.; Hu, Z.; Ye, Y.; Zhang, T.; Li, C.; Zeng, Q.; Zhao, X.; Mai, J.; Sun, Y.; et al. Non-convulsive Status Epilepticus in SEMA6B-Related Progressive Myoclonic Epilepsy: A Case Report With Literature Review. Front. Pediatr. 2022, 10, 859183.
- Hamanaka, K.; Imagawa, E.; Koshimizu, E.; Miyatake, S.; Tohyama, J.; Yamagata, T.; Miyauchi, A.; Ekhilevitch, N.; Nakamura, F.; Kawashima, T.; et al. De Novo Truncating Variants in the Last Exon of SEMA6B Cause Progressive Myoclonic Epilepsy. Am. J. Hum. Genet. 2020, 106, 549–558.
- Chen, Y.; Yang, X.; Yan, X.; Shen, L.; Guo, J.; Xu, Q. A novel SEMA6B variant causes adult-onset progressive myoclonic epilepsy-11 in a Chinese family: A case report and literature review. Front. Genet. 2023, 14, 1110310.
- Lv, X.-J.; Chen, X.; Wang, Y.; Yu, S.; Pang, L.; Huang, C. Aberrant expression of semaphorin 6B affects cell phenotypes in thyroid carcinoma by activating the Notch signalling pathway. Endokrynol. Pol. 2021, 72, 29–36.
- Zhang, S.; Chen, S.; Wang, Y.; Zhan, Y.; Li, J.; Nong, X.; Gao, B. Association of a Novel Prognosis Model with Tumor Mutation Burden and Tumor-Infiltrating Immune Cells in Thyroid Carcinoma. Front. Genet. 2021, 12, 744304.
- Kuznetsova, E.B.; Kekeeva, T.V.; Larin, S.S.; Zemliakova, V.V.; Babenko, O.V.; Nemtsova, M.V.; Zaletaev, D.V.; Strel’nikov, V.V. Novel methylation and expression markers associated with breast cancer. Mol. Biol. 2007, 41, 624–633.
- D’Apice, L.; Costa, V.; Valente, C.; Trovato, M.; Pagani, A.; Manera, S.; Regolo, L.; Zambelli, A.; Ciccodicola, A.; De Berardinis, P. Analysis of SEMA6B gene expression in breast cancer: Identification of a new isoform. Biochim. Biophys. Acta 2013, 1830, 4543–4553.
- Ge, C.; Li, Q.; Wang, L.; Xu, X. The role of axon guidance factor semaphorin 6B in the invasion and metastasis of gastric cancer. J. Int. Med. Res. 2013, 41, 284–292.
- Cui, K.; Bian, X. The microRNA cluster miR-30b/-30d prevents tumor cell switch from an epithelial to a mesenchymal-like phenotype in GBC. Mol. Ther. Methods Clin. Dev. 2021, 20, 716–725.
- Li, T.; Yan, Z.; Wang, W.; Zhang, R.; Gan, W.; Lv, S.; Zeng, Z.; Hou, Y.; Yang, M. SEMA6B Overexpression Predicts Poor Prognosis and Correlates With the Tumor Immunosuppressive Microenvironment in Colorectal Cancer. Front. Mol. Biosci. 2021, 8, 687319.
- Kigel, B.; Rabinowicz, N.; Varshavsky, A.; Kessler, O.; Neufeld, G. Plexin-A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 2011, 118, 4285–4296.
- Collet, P.; Domenjoud, L.; Devignes, M.D.; Murad, H.; Schohn, H.; Dauça, M. The human semaphorin 6B gene is down regulated by PPARs. Genomics 2004, 83, 1141–1150.
- Murad, H.; Collet, P.; Huin-Schohn, C.; Al-Makdissy, N.; Kerjan, G.; Chedotal, A.; Donner, M.; Devignes, M.D.; Becuwe, P.; Schohn, H.; et al. Effects of PPAR and RXR ligands in semaphorin 6B gene expression of human MCF-7 breast cancer cells. Int. J. Oncol. 2006, 28, 977–984.
- Murad, H.; Kasies, F.; Azroony, R.; Alya, G.; Madania, A. Effects of fenofibrate on Semaphorin 6B gene expression in rat skeletal muscle. Mol. Med. Rep. 2011, 4, 575–580.
- Burgaya, F.; Fontana, X.; Martínez, A.; Montolio, M.; Mingorance, A.; Simó, S.; del Río, J.A.; Soriano, E. Semaphorin 6C leads to GSK-3-dependent growth cone collapse and redistributes after entorhino-hippocampal axotomy. Mol. Cell. Neurosci. 2006, 33, 321–334.
- Qu, X.; Wei, H.; Zhai, Y.; Que, H.; Chen, Q.; Tang, F.; Wu, Y.; Xing, G.; Zhu, Y.; Liu, S.; et al. Identification, characterization, and functional study of the two novel human members of the semaphorin gene family. J. Biol. Chem. 2002, 277, 35574–35585.
- Svensson, A.; Libelius, R.; Tågerud, S. Semaphorin 6C expression in innervated and denervated skeletal muscle. J. Mol. Histol. 2008, 39, 5–13.
- Catalano, A.; Caprari, P.; Moretti, S.; Faronato, M.; Tamagnone, L.; Procopio, A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood 2006, 107, 3321–3329.
- Kanth, S.M.; Gairhe, S.; Torabi-Parizi, P. The Role of Semaphorins and Their Receptors in Innate Immune Responses and Clinical Diseases of Acute Inflammation. Front. Immunol. 2021, 12, 672441.
- Naito, Y.; Koyama, S.; Masuhiro, K.; Hirai, T.; Uenami, T.; Inoue, T.; Osa, A.; Machiyama, H.; Watanabe, G.; Sax, N.; et al. Tumor-derived semaphorin 4A improves PD-1-blocking antibody efficacy by enhancing CD8+ T cell cytotoxicity and proliferation. Sci. Adv. 2023, 9, eade0718.
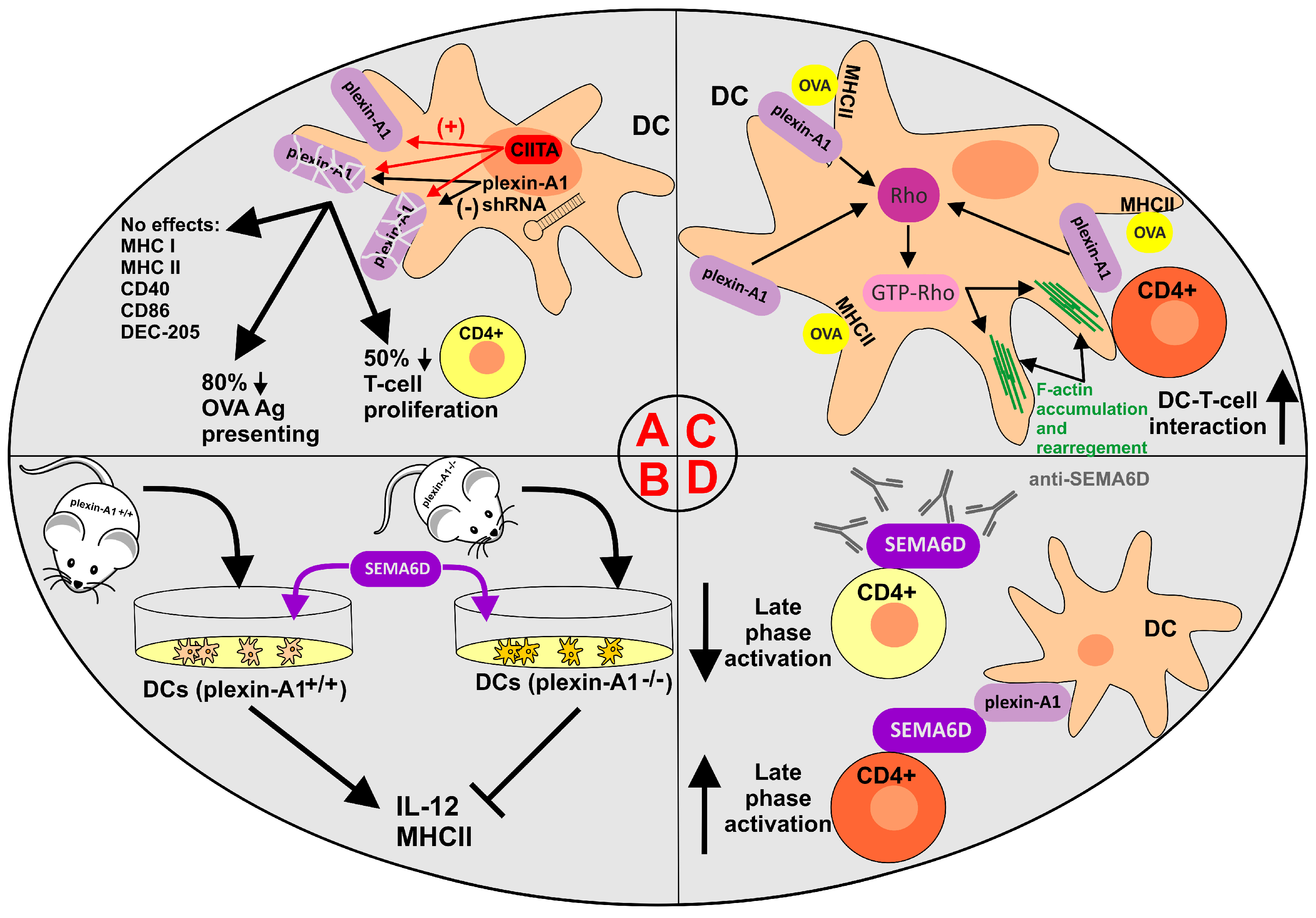
- Wong, A.W.; Brickey, W.J.; Taxman, D.J.; van Deventer, H.W.; Reed, W.; Gao, J.X.; Zheng, P.; Liu, Y.; Li, P.; Blum, J.S.; et al. CIITA-regulated plexin-A1 affects T-cell–dendritic cell interactions. Nat. Immunol. 2003, 4, 891–898.
- Takegahara, N.; Takamatsu, H.; Toyofuku, T.; Tsujimura, T.; Okuno, T.; Yukawa, K.; Mizui, M.; Yamamoto, M.; Prasad, D.V.R.; Suzuki, K.; et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol. 2006, 8, 615–622.
- Eun, S.-Y.; O’Connor, B.P.; Wong, A.W.; van Deventer, H.W.; Taxman, D.J.; Reed, W.; Li, P.; Blum, J.S.; McKinnon, K.P.; Ting, J.P.-Y. Cutting edge: Rho activation and actin polarization are dependent on plexin-A1 in dendritic cells. J. Immunol. 2006, 177, 4271–4275.
- Rodríguez-Fernández, J.L.; Criado-García, O. The Actin Cytoskeleton at the Immunological Synapse of Dendritic Cells. Front. Cell Dev. Biol. 2021, 9, 679500.
- O’Connor, B.P.; Eun, S.-Y.; Ye, Z.; Zozulya, A.L.; Lich, J.D.; Moore, C.B.; Iocca, H.A.; Roney, K.E.; Holl, E.K.; Wu, Q.P.; et al. Semaphorin 6D regulates the late phase of CD4+ T cell primary immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 13015–13020.
- O’Connor, B.P.; Ting, J.P.-Y. The evolving role of semaphorins and plexins in the immune system: Plexin-A1 regulation of dendritic cell function. Immunol. Res. 2008, 41, 217–222.
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17.
- Yamamoto, M.; Suzuki, K.; Okuno, T.; Ogata, T.; Takegahara, N.; Takamatsu, H.; Mizui, M.; Taniguchi, M.; Chédotal, A.; Suto, F.; et al. Plexin-A4 negatively regulates T lymphocyte responses. Int. Immunol. 2008, 20, 413–420.
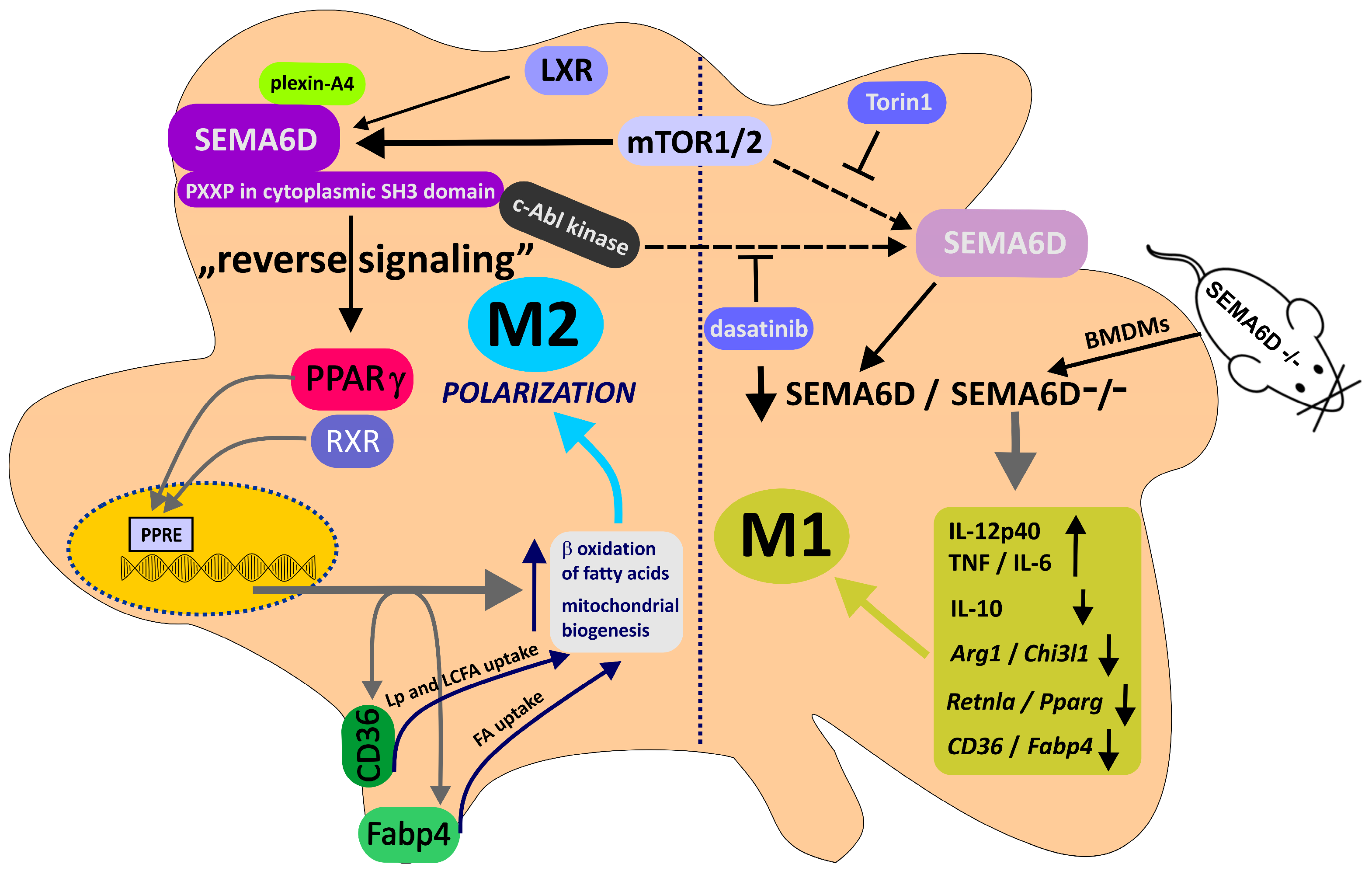
- Kang, S.; Nakanishi, Y.; Kioi, Y.; Okuzaki, D.; Kimura, T.; Takamatsu, H.; Koyama, S.; Nojima, S.; Nishide, M.; Hayama, Y.; et al. Semaphorin 6D reverse signaling controls macrophage lipid metabolism and anti-inflammatory polarization. Nat. Immunol. 2018, 19, 561–570.
- Toyofuku, T.; Zhang, H.; Kumanogoh, A.; Takegahara, N.; Yabuki, M.; Harada, K.; Hori, M.; Kikutani, H. Guidance of myocardial patterning in cardiac development by Sema6D reverse signalling. Nat. Cell Biol. 2004, 6, 1204–1211.
- Locati, M.; Mantovani, A.; Sica, A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013, 120, 163–184.
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604.
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143.
- Sun, Q.; Peng, Y.; Zhao, Q.; Yan, S.; Liu, S.; Yang, Q.; Liu, K.; Rokosh, D.G.; Jiao, K. SEMA6D regulates perinatal cardiomyocyte proliferation and maturation in mice. Dev. Biol. 2019, 452, 1–7.
- Peng, Y.; Song, L.; Li, D.; Kesterson, R.; Wang, J.; Wang, L.; Rokosh, G.; Wu, B.; Wang, Q.; Jiao, K. Sema6D acts downstream of bone morphogenetic protein signalling to promote atrioventricular cushion development in mice. Cardiovasc. Res. 2016, 112, 532–542.
- Papic, N.; Zidovec Lepej, S.; Gorenec, L.; Grgic, I.; Gasparov, S.; Filipec Kanizaj, T.; Vince, A. The association of semaphorins 3C, 5A and 6D with liver fibrosis stage in chronic hepatitis C. PLoS ONE 2018, 13, e0209481.