The function of the circadian cycle is to determine the natural 24 h biological rhythm, which includes physiological, metabolic, and hormonal changes that occur daily in the body. This cycle is controlled by an internal biological clock that is present in the body’s tissues and helps regulate various processes such as sleeping, eating, and others. Interestingly, animal models have provided enough evidence to assume that the alteration in the circadian system leads to the appearance of numerous diseases. Alterations in breathing patterns in lung diseases can modify oxygenation and the circadian cycles; however, the response mechanisms to hypoxia and their relationship with the clock genes are not fully understood. Hypoxia is a condition in which the lack of adequate oxygenation promotes adaptation mechanisms and is related to several genes that regulate the circadian cycles, the latter because hypoxia alters the production of melatonin and brain physiology. Additionally, the lack of oxygen alters the expression of clock genes, leading to an alteration in the regularity and precision of the circadian cycle. In this sense, hypoxia is a hallmark of a wide variety of lung diseases.
- circadian cycle
- lung diseases
- hypoxia
- genes
- idiopathic pulmonary fibrosis
1. Introduction
2. Asthma
3. Chronic Obstructive Pulmonary Disease
4. Lung Cancer
5. Idiopathic Pulmonary Fibrosis
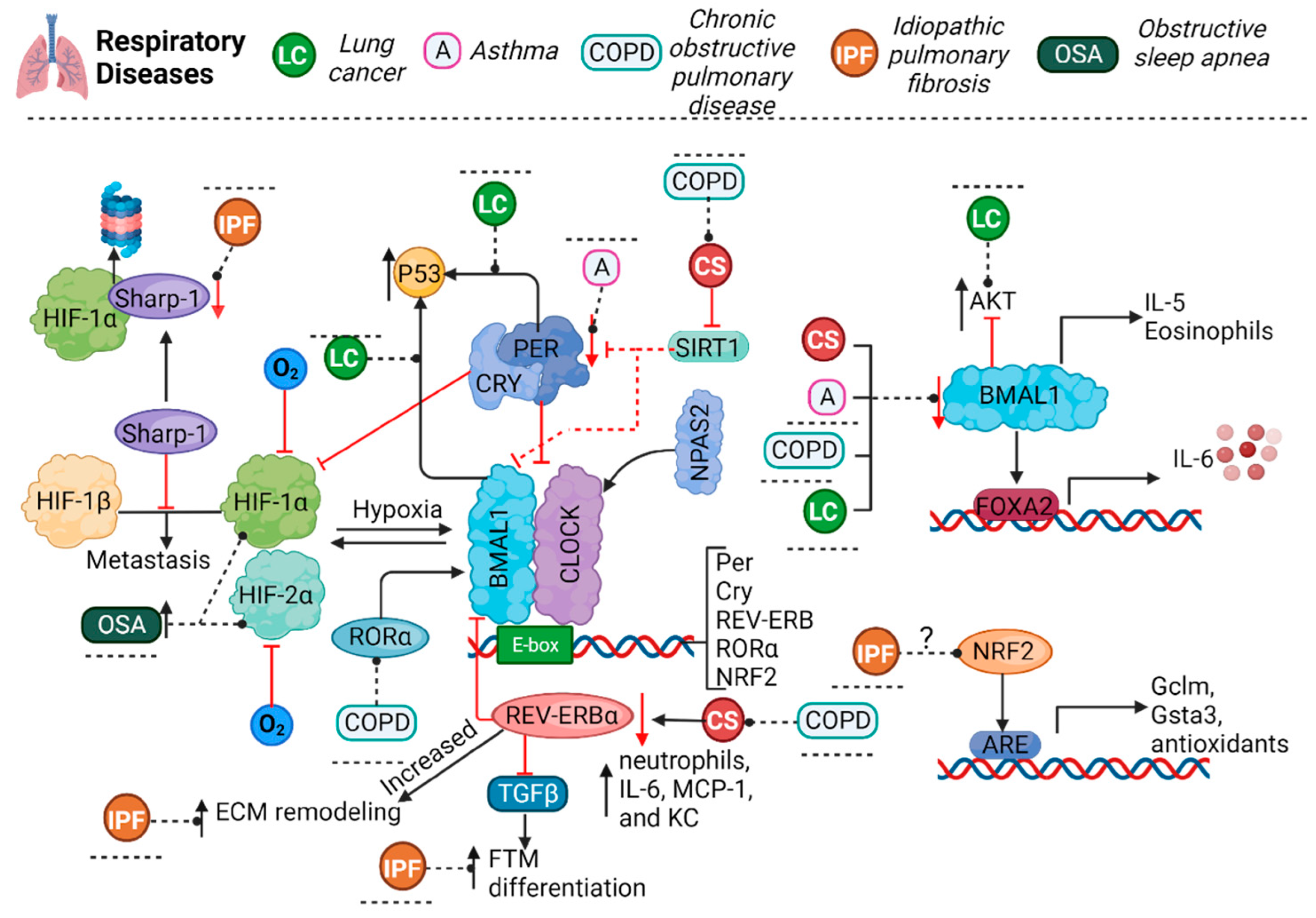
6. Obstructive Sleep Apnea
7. Influenza and COVID-19
This entry is adapted from the peer-reviewed paper 10.3390/cells12232724
References
- Bando, H.; Nishio, T.; Van Der Horst, G.T.J.; Masubuchi, S.; Hisa, Y.; Okamura, H. Vagal Regulation of Respiratory Clocks in Mice. J. Neurosci. 2007, 27, 4359–4365.
- Gibbs, J.E.; Beesley, S.; Plumb, J.; Singh, D.; Farrow, S.; Ray, D.W.; Loudon, A.S. Circadian timing in the lung; a specific role for bronchiolar epithelial cells. Endocrinology. 2009, 150, 268–276.
- Giri, A.; Wang, Q.; Rahman, I.; Sundar, I.K. Circadian molecular clock disruption in chronic pulmonary diseases. Trends Mol. Med. 2022, 28, 513–527.
- Nosal, C.; Ehlers, A.; Haspel, J.A. Why Lungs Keep Time: Circadian Rhythms and Lung Immunity. Annu. Rev. Physiol. 2020, 82, 391–412.
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025.
- Ballard, R.D.; Saathoff, M.C.; Patel, D.K.; Kelly, P.L.; Martin, R.J. Effect of sleep on nocturnal bronchoconstriction and ventilatory patterns in asthmatics. J. Appl. Physiol. 1989, 67, 243–249.
- Scheer, F.A.J.L.; Hilton, M.F.; Evoniuk, H.L.; Shiels, S.A.; Malhotra, A.; Sugarbaker, R.; Ayers, R.T.; Israel, E.; Massaro, A.F.; Shea, S.A. The Endogenous Circadian System Worsens Asthma at Night Independent of Sleep and Other Daily Behavioral or Environmental Cycles. Proc. Natl. Acad. Sci. USA 2021, 118, e2018486118.
- Sutherland, E.R.; Ellison, M.C.; Kraft, M.; Martin, R.J. Elevated serum melatonin is associated with the nocturnal worsening of asthma. J. Allergy Clin. Immunol. 2003, 112, 513–517.
- Jönsson, E.; Mossberg, B. Impairment of ventilatory function by supine posture in asthma. Eur. J. Respir. Dis. 1984, 65, 496–503.
- Chen, W.Y.; Chai, H. Airway cooling and nocturnal asthma. Chest 1982, 81, 675–680.
- Maidstone, R.J.; Turner, J.; Vetter, C.; Dashti, H.S.; Saxena, R.; Scheer, F.A.J.L.; Shea, S.A.; Kyle, S.D.; Lawlor, D.A.; Loudon, A.S.I. Night Shift Work Is Associated with an Increased Risk of Asthma. Thorax 2021, 76, 53–60.
- Landstra, A.M.; Postma, D.S.; Boezen, H.M.; van Aalderen, W.M. R ole of serum cortisol levels in children with asthma. Am. J. Respir. Crit. Care Med. 2002, 165, 708–712.
- Sutherland, E.R.; Ellison, M.C.; Kraft, M.; Martin, R.J. Altered Pituitary-adrenal Interaction in Nocturnal Asthma. J. Allergy Clin. Immunol. 2003, 112, 52–57.
- Ehlers, A.; Xie, W.; Agapov, E.; Brown, S.; Steinberg, D.; Tidwell, R.; Sajol, G.; Schutz, R.; Weaver, R.; Yu, H. BMAL1 Links the Circadian Clock to Viral Airway Pathology and Asthma Phenotypes. Mucosal Immunol. 2018, 11, 97–111.
- Chen, H.-C.; Chen, Y.-C.; Wang, T.-N.; Fang, W.-F.; Chang, Y.-C.; Chen, Y.-M.; Chen, I.-Y.; Lin, M.-C.; Yang, M.-Y. Disrupted Expression of Circadian Clock Genes in Patients with Bronchial Asthma. J. Asthma Allergy 2021, 14, 371–380.
- Tang, L.; Liu, L.; Sun, X.; Hu, P.; Zhang, H.; Wang, B.; Zhang, X.; Jiang, J.; Zhao, X.; Shi, X. BMAL1/FOXA2-induced rhythmic fluctuations in IL-6 contribute to nocturnal asthma attacks. Front. Immunol. 2022, 13, 947067.
- Zasłona, Z.; Case, S.; Early, J.O.; Lalor, S.J.; McLoughlin, R.M.; Curtis, A.M.; O’Neill, L.A.J. The circadian protein BMAL1 in myeloid cells is a negative regulator of allergic asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L855–L860.
- Partridge, M.R.; Karlsson, N.; Small, I.R. Patient insight into the impact of chronic obstructive pulmonary disease in the morning: An internet survey. Curr. Med. Res. Opin. 2009, 25, 2043–2048.
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette Smoke-induced Autophagy Impairment Accelerates Lung Aging, Copd-emphysema Exacerbations and Pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87.
- Casale, R.; Pasqualetti, P. Cosinor analysis of circadian peak expiratory flow variability in normal subjects, passive smokers, heavy smokers, patients with chronic obstructive pulmonary disease and patients with interstitial lung disease. Respir. Int. Rev. Thorac. Dis. 1997, 64, 251–256.
- Dawkins, K.D.; Muers, M.F. Diurnal Variation in Airflow Obstruction in Chronic Bronchitis. Thorax 1981, 36, 618–621.
- Calverley, P.M.A. Effect of Tiotropium Bromide on Circadian Variation in Airflow Limitation in Chronic Obstructive Pulmonary Disease. Thorax 2003, 58, 855–860.
- Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian Molecular Clock in Lung Pathophysiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1056–L1075.
- Hwang, J.W.; Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biology 2014, 28, 176–194.
- Sundar, I.K.; Rashid, K.; Sellix, M.T.; Rahman, I. The Nuclear Receptor and Clock Gene Rev-erbα Regulates Cigarette Smoke-induced Lung Inflammation. Biochem. Biophys. Res. Commun. 2017, 493, 1390–1395.
- Wang, Q.; Sundar, I.K.; Lucas, J.H.; Muthumalage, T.; Rahman, I. Molecular Clock Rev-erbα Regulates Cigarette Smoke–induced Pulmonary Inflammation and Epithelial-mesenchymal Transition. JCI Insight 2021, 6, e145200.
- Shi, Y.; Cao, J.; Gao, J.; Zheng, L.; Goodwin, A.; An, C.H.; Patel, A.; Lee, J.S.; Duncan, S.R.; Kaminski, N. Retinoic Acid–related Orphan Receptor-α Is Induced in the Setting of DNA Damage and Promotes Pulmonary Emphysema. Am. J. Respir. Crit. Care Med. 2012, 186, 412–419.
- Yao, H.; Sundar, I.K.; Huang, Y.; Gerloff, J.; Sellix, M.T.; Sime, P.J.; Rahman, I. Disruption of Sirtuin 1–mediated Control of Circadian Molecular Clock and Inflammation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2015, 53, 782–792.
- Li, L.; Zhang, M.; Zhao, C.; Cheng, Y.; Liu, C.; Shi, M. Circadian Clock Gene Clock-bmal1 Regulates Cellular Senescence in Chronic Obstructive Pulmonary Disease. BMC Pulm. Med. 2022, 22, 435.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424.
- Larsson, S.C.; Carter, P.; Kar, S.; Vithayathil, M.; Mason, A.M.; Michaëlsson, K.; Burgess, S. Smoking, Alcohol Consumption, and Cancer: A Mendelian Randomisation Study in UK Biobank and International Genetic Consortia Participants. PLOS Med. 2020, 17, e1003178.
- Gandini, S.; Botteri, E.; Iodice, S.; Boniol, M.; Lowenfels, A.B.; Maisonneuve, P.; Boyle, P. Tobacco Smoking and Cancer: A Meta-analysis. Int. J. Cancer 2008, 122, 155–164.
- Aredo, J.V.; Luo, S.J.; Gardner, R.M.; Sanyal, N.; Choi, E.; Hickey, T.P.; Riley, T.L.; Huang, W.-Y.; Kurian, A.W.; Leung, A.N. Tobacco Smoking and Risk of Second Primary Lung Cancer. J. Thorac. Oncol. 2021, 16, 968–979.
- Wang, J.; Tang, H.; Duan, Y.; Yang, S.; An, J. Association Between Sleep Traits and Lung Cancer: A Mendelian Randomization Study. J. Immunol. Res. 2021, 2021, 1893882.
- Huang, B.-H.; Duncan, M.J.; Cistulli, P.A.; Nassar, N.; Hamer, M.; Stamatakis, E. Sleep and Physical Activity in Relation to All-cause, Cardiovascular Disease and Cancer Mortality Risk. Br. J. Sport. Med. 2022, 56, 718–724.
- Stone, C.R.; Haig, T.R.; Fiest, K.M.; Mcneil, J.; Brenner, D.R.; Friedenreich, C.M. The Association between Sleep Duration and Cancer-specific Mortality: A Systematic Review and Meta-analysis. Cancer Causes Control 2019, 30, 501–525.
- Xie, J.; Zhu, M.; Ji, M.; Fan, J.; Huang, Y.; Wei, X.; Jiang, X.; Xu, J.; Yin, R.; Wang, Y.; et al. Relatsh Between Sleep Trait Lung Cancer Risk: A Prospect. Cohort Study UK Biobank. Sleep 2021, 44, zsab089.
- Huo, Z.; Ge, F.; Li, C.; Cheng, H.; Lu, Y.; Wang, R.; Wen, Y.; Yue, K.; Pan, Z.; Peng, H.; et al. Genetically predicted insomnia and lung cancer risk: A Mendelian randomization study. Sleep Med. 2021, 87, 183–190.
- Cao, Q.; Zhang, Q.; Li, X.C.; Ren, C.F.; Qiang, Y. Impact of sleep status on lung adenocarcinoma risk: A prospective cohort study. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 7641–7648.
- Zeng, Z.L.; Wu, M.W.; Sun, J.; Sun, Y.L.; Cai, Y.C.; Huang, Y.J.; Xian, L.J. Effects of the biological clock gene Bmal1 on tumour growth and anti-cancer drug activity. J. Biochem. 2010, 148, 319–326.
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494.
- Qiu, M.; Chen, Y.B.; Jin, S.; Fang, X.F.; He, X.X.; Xiong, Z.F.; Yang, S.L. Research on circadian clock genes in non-small-cell lung carcinoma. Chronobiol. Int. 2019, 36, 739–750.
- Vasu, V.T.; Cross, C.E.; Gohil, K. Nr1d1, an important circadian pathway regulatory gene, is suppressed by cigarette smoke in murine lungs. Integr. Cancer Ther. 2009, 8, 321–328.
- Jiang, W.; Zhao, S.; Jiang, X.; Zhang, E.; Hu, G.; Hu, B.; Zheng, P.; Xiao, J.; Lu, Z.; Lu, Y.; et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 2016, 371, 314–325.
- Gotoh, T.; Vila-Caballer, M.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The Circadian Factor Period 2 Modulates P53 Stability and Transcriptional Activity in Unstressed Cells. Mol. Biol. Cell 2014, 25, 3081–3093.
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-driven Experimental Approach Reveals the Complex Regulatory Distribution of P53 by the Circadian Factor Period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521.
- Gotoh, T.; Vila-Caballer, M.; Liu, J.; Schiffhauer, S.; Finkielstein, C.V. Association of the Circadian Factor Period 2 to P53 Influences P53′s Function in Dna-damage Signaling. Mol. Biol. Cell 2015, 26, 359–372.
- Zhanfeng, N.; Chengquan, W.; Hechun, X.; Jun, W.; Lijian, Z.; Dede, M.; Wenbin, L.; Lei, Y. Period2 Downregulation Inhibits Glioma Cell Apoptosis by Activating the MDM2-TP53 Pathway. Oncotarget 2016, 7, 27350–27362.
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The Circadian Gene Period2 Plays an Important Role in Tumor Suppression and DNA Damage Response in Vivo. Cell 2002, 111, 41–50.
- Jung, C.-H.; Kim, E.M.; Park, J.K.; Hwang, S.-G.; Moon, S.-K.; Kim, W.-J.; Um, H.-D. Bmal1 Suppresses Cancer Cell Invasion by Blocking the Phosphoinositide 3-kinase-akt-mmp-2 Signaling Pathway. Oncol. Rep. 2013, 29, 2109–2113.
- Iksen; Pothongsrisit, S.; Pongrakhananon, V. Targeting the Pi3k/akt/mtor Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100.
- Bastani, S.; Akbarzadeh, M.; Rastgar Rezaei, Y.; Farzane, A.; Nouri, M.; Mollapour Sisakht, M.; Fattahi, A.; Akbarzadeh, M.; Reiter, R.J. Melatonin as a Therapeutic Agent for the Inhibition of Hypoxia-induced Tumor Progression: A Description of Possible Mechanisms Involved. Int. J. Mol. Sci. 2021, 22, 10874.
- Zhao, H.; Wu, Q.-Q.; Cao, L.-F.; Qing, H.-Y.; Zhang, C.; Chen, Y.-H.; Wang, H.; Liu, R.-R.; Xu, D.-X. Melatonin Inhibits Endoplasmic Reticulum Stress and Epithelial-mesenchymal Transition During Bleomycin-induced Pulmonary Fibrosis in Mice. PLoS ONE 2014, 9, e97266.
- Shen, H.; Cook, K.; Gee, H.E.; Hau, E. Hypoxia, Metabolism, and the Circadian Clock: New Links to Overcome Radiation Resistance in High-grade Gliomas. J. Exp. Clin. Cancer Res. 2020, 39, 129.
- Balsara, B.R.; Pei, J.; Mitsuuchi, Y.; Page, R.; Klein-Szanto, A.; Wang, H.; Unger, M.; Testa, J.R. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 2004, 25, 2053–2059.
- Scheffler, M.; Bos, M.; Gardizi, M.; König, K.; Michels, S.; Fassunke, J.; Heydt, C.; Künstlinger, H.; Ihle, M.; Ueckeroth, F. PIK3CA Mutations in Non-small Cell Lung Cancer (NSCLC): Genetic Heterogeneity, Prognostic Impact and Incidence of Prior Malignancies. Oncotarget 2015, 6, 1315–1326.
- Zhao, D.; Dong, Y.; Duan, M.; He, D.; Xie, Q.; Peng, W.; Cui, W.; Jiang, J.; Cheng, Y.; Zhang, H. Circadian Gene ARNTL Initiates Circgucy1a2 Transcription to Suppress Non-small Cell Lung Cancer Progression via Mir-200c-3p/pten Signaling. J. Exp. Clin. Cancer Res. 2023, 42, 229.
- Rossner, M.J.; Oster, H.; Wichert, S.P.; Reinecke, L.; Wehr, M.C.; Reinecke, J.; Eichele, G.; Taneja, R.; Nave, K.-A. Disturbed Clockwork Resetting in Sharp-1 and Sharp-2 Single and Double Mutant Mice. PLoS ONE 2008, 3, e2762.
- Liu, Q.; Wu, Y.; Seino, H.; Haga, T.; Yoshizawa, T.; Morohashi, S.; Kijima, H. Correlation between DEC1/DEC2 and Epithelial-mesenchymal Transition in Human Prostate Cancer PC-3 Cells. Mol. Med. Rep. 2018, 18, 3859–3865.
- Wu, Y.; Sato, H.; Suzuki, T.; Yoshizawa, T.; Morohashi, S.; Seino, H.; Kawamoto, T.; Fujimoto, K.; Kato, Y.; Kijima, H. Involvement of C-myc in the Proliferation of MCF-7 Human Breast Cancer Cells Induced by Bhlh Transcription Factor DEC2. Int. J. Mol. Med. 2015, 35, 815–820.
- Amelio, I.; Melino, G. The “sharp” blade against Hif-mediated Metastasis. Cell Cycle 2012, 11, 4530–4535.
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S. SHARP1 Suppresses Breast Cancer Metastasis by Promoting Degradation of Hypoxia-inducible Factors. Nature 2012, 487, 380–384.
- Liao, Y.; Lu, W.; Che, Q.; Yang, T.; Qiu, H.; Zhang, H.; He, X.; Wang, J.; Qiu, M.; Zou, Y. SHARP1 Suppresses Angiogenesis of Endometrial Cancer by Decreasing Hypoxia-inducible Factor-1α Level. PLoS ONE 2014, 9, e99907.
- Piccolo, S.; Enzo, E.; Montagner, M. p63, Sharp1, and HIFs: Master regulators of metastasis in triple-negative breast cancer. Cancer Res. 2013, 73, 4978–4981.
- Falvella, F.S.; Colombo, F.; Spinola, M.; Campiglio, M.; Pastorino, U.; Dragani, T.A. BHLHB3: A Candidate Tumor Suppressor in Lung Cancer. Oncogene 2008, 27, 3761–3764.
- Gallo, C.; Fragliasso, V.; Donati, B.; Torricelli, F.; Tameni, A.; Piana, S.; Ciarrocchi, A. The Bhlh Transcription Factor DEC1 Promotes Thyroid Cancer Aggressiveness by the Interplay with NOTCH1. Cell Death Dis. 2018, 9, 871.
- Liao, Y.; He, X.; Qiu, H.; Che, Q.; Wang, F.; Lu, W.; Chen, Z.; Qiu, M.; Wang, J.; Wang, H. Suppression of the Epithelial-mesenchymal Transition by SHARP1 Is Linked to the NOTCH1 Signaling Pathway in Metastasis of Endometrial Cancer. BMC Cancer 2014, 14, 487.
- Liu, J.-J.; Chung, T.-K.; Li, J.; Taneja, R. Sharp-1 Modulates the Cellular Response to DNA Damage. FEBS Lett. 2010, 584, 619–624.
- Nakamura, H.; Tanimoto, K.; Hiyama, K.; Yunokawa, M.; Kawamoto, T.; Kato, Y.; Yoshiga, K.; Poellinger, L.; Hiyama, E.; Nishiyama, M. Human Mismatch Repair Gene, MLH1, Is Transcriptionally Repressed by the Hypoxia-inducible Transcription Factors, DEC1 and DEC2. Oncogene 2008, 27, 4200–4209.
- Liu, Y.; Wang, L.; Lin, X.-Y.; Wang, J.; Yu, J.-H.; Miao, Y.; Wang, E.-H. The Transcription Factor DEC1 (BHLHE40/STRA13/SHARP-2) Is Negatively Associated with TNM Stage in Non-small-cell Lung Cancer and Inhibits the Proliferation through Cyclin D1 in A549 and BE1 Cells. Tumor Biol. 2013, 34, 1641–1650.
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824.
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary Fibrosis: Patterns and Perpetrators. J. Clin. Investig. 2012, 122, 2756–2762.
- Kim, R.; Meyer, K.C. Therapies for interstitial lung disease: Past, present and future. Ther. Adv. Respir. Dis. 2008, 2, 319–338.
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77.
- Gharaee-Kermani, M.; Hu, B.; Phan, S.H.; Gyetko, M.R. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: Focus on TGFbeta signaling and the myofibroblast. Curr. Med. Chem. 2009, 16, 1400–1417.
- Khalil, N.; Xu, Y.D.; O’Connor, R.; Duronio, V. Proliferation of Pulmonary Interstitial Fibroblasts Is Mediated by Transforming Growth Factor-β1-induced Release of Extracellular Fibroblast Growth Factor-2 and Phosphorylation of P38 MAPK and JNK. J. Biol. Chem. 2005, 280, 43000–43009.
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961.
- Zhang, K.; Flanders, K.C.; Phan, S.H. Cellular localization of transforming growth factor-beta expression in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 1995, 147, 352–361.
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated Gene Transfer of Active Transforming Growth Factor-beta1 Induces Prolonged Severe Fibrosis in Rat Lung. J. Clin. Investig. 1997, 100, 768–776.
- Warshamana, G.S.; Pociask, D.A.; Fisher, K.J.; Liu, J.-Y.; Sime, P.J.; Brody, A.R. Titration of Non-replicating Adenovirus as a Vector for Transducing Active Tgf-β1 Gene Expression Causing Inflammation and Fibrogenesis in the Lungs of C57BL/6 Mice. Int. J. Exp. Pathol. 2002, 83, 183–202.
- Gibbs, J.; Ince, L.; Matthews, L.; Mei, J.; Bell, T.; Yang, N.; Saer, B.; Begley, N.; Poolman, T.; Pariollaud, M. An Epithelial Circadian Clock Controls Pulmonary Inflammation and Glucocorticoid Action. Nat. Med. 2014, 20, 919–926.
- Cunningham, P.S.; Meijer, P.; Nazgiewicz, A.; Anderson, S.G.; Borthwick, L.A.; Bagnall, J.; Kitchen, G.B.; Lodyga, M.; Begley, N.; Venkateswaran, R.V. The Circadian Clock Protein Reverbα Inhibits Pulmonary Fibrosis Development. Proc. Natl. Acad. Sci. USA 2020, 117, 1139–1147.
- Romero, Y.; Balderas-Martínez, Y.I.; Vargas-Morales, M.A.; Castillejos-López, M.; Vázquez-Pérez, J.A.; Calyeca, J.; Torres-Espíndola, L.M.; Patiño, N.; Camarena, A.; Carlos-Reyes, Á. Effect of Hypoxia in the Transcriptomic Profile of Lung Fibroblasts from Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 3014.
- Patel, S.R. Obstructive Sleep Apnea. Ann. Intern. Med. 2019, 171, ITC81–ITC96.
- Rundo, J.V. Obstructive Sleep Apnea Basics. Clevel. Clin. J. Med. 2019, 86, 2–9.
- Dempsey, J.A. Central Sleep Apnea: Misunderstood and Mistreated! F1000Research 2019, 8, 981.
- Ishikawa, O.; Oks, M. Central Sleep Apnea. Clin. Geriatr. Med. 2021, 37, 469–481.
- Şenel, G.B.; Aliş, C.; Karadeniz, D. Are We Underestimating the Central Components of the Mixed Apneas?-A Hypothesis for Revised Scoring. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2023, 40, 165–172.
- Semelka, M.; Wilson, J.; Floyd, R. Diagnosis and Treatment of Obstructive Sleep Apnea in Adults. Am. Fam. Physician 2016, 94, 355–360.
- Qaseem, A.; Dallas, P.; Owens, D.K.; Starkey, M.; Holty, J.E.; Shekelle, P.; Clinical Guidelines Committee of the American College of Physicians. Diagnosis of obstructive sleep apnea in adults: A clinical practice guideline from the American College of Physicians. Ann. Intern. Med. 2014, 161, 210–220.
- Walia, R.; Achilefu, A.; Crawford, S.; Jain, V.; Wigley, S.D.; McCarthy, L.H. Are at-home sleep studies performed using portable monitors (PMs) as effective at diagnosing obstructive sleep apnea (OSA) in adults as sleep laboratory-based polysomnography (PSG)? J. Okla. State Med. Assoc. 2014, 107, 642–644.
- Epstein, L.J.; Kristo, D.; Strollo, P.J., Jr.; Friedman, N.; Malhotra, A.; Patil, S.P.; Ramar, K.; Rogers, R.; Schwab, R.J.; Weaver, E.M.; et al. Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J. Clin. Sleep Med. 2009, 5, 263–276.
- Bonsignore, M.R.; Baiamonte, P.; Mazzuca, E.; Castrogiovanni, A.; Marrone, O. Obstructive Sleep Apnea and Comorbidities: A Dangerous Liaison. Multidiscip. Respir. Med. 2019, 14, 8.
- Marin-Oto, M.; Vicente, E.E.; Marin, J.M. Long Term Management of Obstructive Sleep Apnea and Its Comorbidities. Multidiscip. Respir. Med. 2019, 14, 21.
- Coste, O.; Beaumont, M.; Batéjat, D.; Beers, P.V.; Touitou, Y. Prolonged mild hypoxia modifies human circadian core body temperature and may be associated with sleep disturbances. Chronobiol. Int. 2004, 21, 419–433.
- Coste, O.; Beaumont, M.; Batéjat, D.; Beers, P.V.; Charbuy, H.; Touitou, Y. Hypoxic Depression of Melatonin Secretion After Simulated Long Duration Flights in Man. J. Pineal Res. 2004, 37, 1–10.
- Coste, O.; Beers, P.V.; Bogdan, A.; Charbuy, H.; Touitou, Y. Hypoxic alterations of cortisol circadian rhythm in man after simulation of a long duration flight. Steroids 2005, 70, 803–810.
- Wu, Y.; Tang, D.; Liu, N.; Xiong, W.; Huang, H.; Li, Y.; Ma, Z.; Zhao, H.; Chen, P.; Qi, X. Reciprocal Regulation Between the Circadian Clock and Hypoxia Signaling at the Genome Level in Mammals. Cell Metab. 2017, 25, 73–85.
- Yu, C.; Yang, S.-L.; Fang, X.; Jiang, J.-X.; Sun, C.-Y.; Huang, T. Hypoxia Disrupts the Expression Levels of Circadian Rhythm Genes in Hepatocellular Carcinoma. Mol. Med. Rep. 2015, 11, 4002–4008.
- Yang, M.Y.; Lin, P.W.; Lin, H.C.; Lin, P.M.; Chen, I.Y.; Friedman, M.; Hung, C.F.; Salapatas, A.M.; Lin, M.C.; Lin, S.F. Alternations of Circadian Clock Genes Expression and Oscillation in Obstructive Sleep Apnea. J. Clin. Med. 2019, 8, 1634.
- Pellegrino, R.; Kavakli, I.H.; Goel, N.; Cardinale, C.J.; Dinges, D.F.; Kuna, S.T.; Maislin, G.; Van Dongen, H.P.; Tufik, S.; Hogenesch, J.B.; et al. A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep 2014, 37, 1327–1336.
- Clayville, L.R. Influenza update: A review of currently available vaccines. P T A Peer-Rev. J. Formul. Manag. 2011, 36, 659–684.
- Gaitonde, D.Y.; Moore, F.C.; Morgan, M.K. Influenza: Diagnosis and Treatment. Am. Fam. Physician. 2019, 100, 751–758.
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A brief review of influenza virus infection. J. Med. Virol. 2021, 93, 4638–4646.
- Nakagawa, H.; Noma, H.; Kotake, O.; Motohashi, R.; Yasuda, K.; Shimura, M. Optic Neuritis and Acute Anterior Uveitis Associated with Influenza A Infection: A Case Report. Int. Med. Case Rep. J. 2017, 10, 1–5.
- Dharmapalan, D. Influenza. Indian J. Pediatr. 2020, 87, 828–832.
- Sundar, I.K.; Ahmad, T.; Yao, H.; Hwang, J.-W.; Gerloff, J.; Lawrence, B.P.; Sellix, M.T.; Rahman, I. Influenza A Virus-dependent Remodeling of Pulmonary Clock Function in a Mouse Model of COPD. Sci. Rep. 2015, 5, 9927.
- Zhang, Z.; Hunter, L.; Wu, G.; Maidstone, R.; Mizoro, Y.; Vonslow, R.; Fife, M.; Hopwood, T.; Begley, N.; Saer, B. Genome-wide Effect of Pulmonary Airway Epithelial Cell–specific Bmal1 Deletion. FASEB J. 2019, 33, 6226–6238.
- Zhuang, X.; Magri, A.; Hill, M.; Lai, A.G.; Kumar, A.; Rambhatla, S.B.; Donald, C.L.; Lopez-Clavijo, A.F.; Rudge, S.; Pinnick, K. The Circadian Clock Components BMAL1 and Rev-erbα Regulate Flavivirus Replication. Nat. Commun. 2019, 10, 377.
- Borrmann, H.; Davies, R.; Dickinson, M.; Pedroza-Pacheco, I.; Schilling, M.; Vaughan-Jackson, A.; Magri, A.; James, W.; Balfe, P.; Borrow, P. Pharmacological Activation of the Circadian Component REV-ERB Inhibits HIV-1 Replication. Sci. Rep. 2020, 10, 13271.
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C. Clinical Characteristics of Coronavirus Disease 2019 in China. New Engl. J. Med. 2020, 382, 1708–1720.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl. J. Med. 2020, 382, 727–733.
- Morin, C.M.; Carrier, J.; Bastien, C.; Godbout, R. Sleep and Circadian Rhythm in Response to the COVID-19 Pandemic. Can. J. Public Health 2020, 111, 654–657.
- Lai, J.; Ma, S.; Wang, Y.; Cai, Z.; Hu, J.; Wei, N.; Wu, J.; Du, H.; Chen, T.; Li, R. Factors Associated with Mental Health Outcomes Among Health Care Workers Exposed to Coronavirus Disease 2019. JAMA Netw. Open 2020, 3, e203976.
- Irwin, M.R. Sleep and Inflammation: Partners in Sickness and in Health. Nat. Rev. Immunol. 2019, 19, 702–715.
- Anderson, G.; Reiter, R.J. Melatonin: Roles in Influenza, COVID-19, and Other Viral Infections. Rev. Med. Virol. 2020, 30, e2109.
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W. The Nuclear Receptor Rev-erbα Mediates Circadian Regulation of Innate Immunity Through Selective Regulation of Inflammatory Cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587.
- Haspel, J.A.; Anafi, R.; Brown, M.K.; Cermakian, N.; Depner, C.; Desplats, P.; Gelman, A.E.; Haack, M.; Jelic, S.; Kim, B.S. Perfect Timing: Circadian Rhythms, Sleep, and Immunity—An NIH Workshop Summary. JCI Insight 2020, 5, e131487.
- Costantini, C.; Renga, G.; Sellitto, F.; Borghi, M.; Stincardini, C.; Pariano, M.; Zelante, T.; Chiarotti, F.; Bartoli, A.; Mosci, P.; et al. Microbes Era Circadian Medicine. Front. Cell. Infect. Microbiol. 2020, 10, 30.
- Zhuang, X.; Rambhatla, S.B.; Lai, A.G.; Mckeating, J.A. Interplay between Circadian Clock and Viral Infection. J. Mol. Med. 2017, 95, 1283–1289.
- Mazzoccoli, G.; Vinciguerra, M.; Carbone, A.; Relógio, A. The Circadian Clock, the Immune System, and Viral Infections: The Intricate Relationship between Biological Time and Host-virus Interaction. Pathogens 2020, 9, 83.
- Pariollaud, M.; Gibbs, J.E.; Hopwood, T.W.; Brown, S.; Begley, N.; Vonslow, R.; Poolman, T.; Guo, B.; Saer, B.; Jones, D.H. Circadian Clock Component Rev-erbα Controls Homeostatic Regulation of Pulmonary Inflammation. J. Clin. Investig. 2018, 128, 2281–2296.
- Perry, M.G.; Kirwan, J.R.; Jessop, D.S.; Hunt, L.P. Overnight Variations in Cortisol, Interleukin 6, Tumour Necrosis Factor A and Other Cytokines in People with Rheumatoid Arthritis. Ann. Rheum. Dis. 2009, 68, 63–68.