Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Non-alcoholic fatty liver disease (NAFLD) affects up to a quarter of the adult population in many developed and developing countries. This spectrum of liver disease ranges from simple steatosis to non-alcoholic steatohepatitis (NASH) and cirrhosis. The incidence of NASH is projected to increase by up to 56% in the future. There is growing epidemiological evidence that NAFLD has become the fastest-growing cause of hepatocellular carcinoma (HCC) in industrialized countries. The annual incidence of HCC varies between patients with NASH cirrhosis and patients with noncirrhotic NAFLD.
- NAFLD
- NASH
- HCC
- SNP
1. Introduction
The main risk factor for the development of HCC is the presence of liver cirrhosis [1]. Other major risk factors among patients with NAFLD are obesity, diabetes, and dyslipidemia [2]. Emerging data also implicate gut dysbiosis and inflammation as additional key risk factors for HCC development in patients with NAFLD. However, there are other demographic, metabolic, genetic, and environmental factors that have been associated with the development of NAFLD [3] (Figure 1).
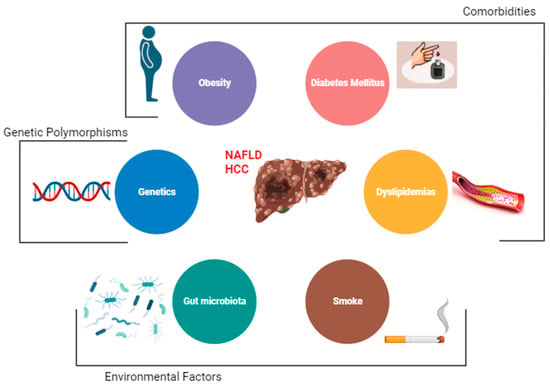
Figure 1. Risk factors for hepatocellular carcinoma (HCC) among NAFLD patients.
2. Obesity
Obesity and NAFLD/NASH are becoming the leading contributing factors to the rising incidence of HCC [4]. Obesity is a major driver of NAFLD and NASH, [3] and it is associated with a 2–3-fold increased risk of HCC [5]. Notably, obesity itself is an independent risk factor for the onset and development of HCC.
In a retrospective cohort study of 271,906 patients with diagnosed NAFLD, it has been shown that 8.38% subjects developed cirrhosis, and 253 were diagnosed with HCC (0.09% of patients with NAFLD and 1.11% with cirrhosis, respectively) [4].
In another analysis, which analyzed 25,337 HCC patients from a total of 26 prospective studies, overweight and obesity increased the risk of HCC by 18% and 83%, respectively, regardless of gender and geographical location. The incidence was found to be higher in men than in women, but this could be derived from the different distribution of adipose tissue with a higher percentage of visceral obesity in men [6].
The exact biological mechanisms linking weight gain and HCC have not yet been fully elucidated; however, it can be assumed that the development of NAFLD and NASH represent milestones. In fact, obesity and the resulting insulin resistance favor the release of pro-inflammatory cytokines such as TNFα and IL-6, responsible for the development of hepatic steatosis, inflammation, and the onset of HCC [7].
Considering only abdominal obesity, rather than the BMI index, an even closer relationship emerges between obesity, NAFLD, and HCC; in fact, in a detailed analysis of the risk of developing HCC, waist and hip circumference, waist-to-hip ratio (WHR), and waist-to-height ratio (WHtR) have been described positively associated with risk of HCC. In particular, WHtR showed the strongest association with HCC [8].
These parameters appear to be more predictive and worthy of greater attention as they identify more precisely the visceral fat, responsible for the basic metabolic and inflammatory activity and effector of the damage.
3. Diabetes Mellitus
The association between type 2 diabetes mellitus (T2DM) and NAFLD is strongly supported by several studies [9][10][11].
A study of Mayo Clinic and UNOS on 6984 patients demonstrated that diabetes is involved not only in the onset of NASH (as part of the metabolic syndrome) but also in the progression of liver disease and in the onset of cirrhosis following NAFLD [11]. Diabetes promotes hepatocarcinogenesis by constituting a chronic inflammatory state that favors the release of proinflammatory cytokines (leptin and TNF-α) and the formation of oxygen free radicals (ROS). ROS cause genomic instability, promote cell differentiation, and inhibit apoptosis. In addition, diabetes is associated with hyperinsulinemia and IGF-1-activated growth factors. Insulin and IGF-1 act on the PI3K/AKT and MAPK molecular pathways. The activation of PI3K/AKT leads to inhibition of apoptosis and the increase in growth and cell survival by signaling on cyclin D1, p53, and Mtor; the activation of MAPK stimulates the transcription of proto-oncogenes, explaining the high incidence of HCC in diabetic patients [4].
A retrospective analysis on patients with and without DM demonstrated that diabetics were older, predominantly female, had metabolic syndrome, and had NAFLD as the underlying etiology. Moreover, in a median follow-up period of 6 months among 156 patients without cirrhosis, a higher proportion (43% vs. 27%) of diabetics than non-diabetics developed cirrhosis. Similarly, over a median follow-up of 3 years, among 359 patients with cirrhosis at or during follow-up, a higher proportion of diabetics (22% vs. 5%) developed HCC. Interestingly, oral antidiabetic drugs (e.g., Metformin or Thiazolidinedones), because of their intrinsic mechanism of action which counteracts insulin resistance, have been demonstrated to be more effective in controlling the progression of liver damage when compared with insulin therapy [10].
4. Dyslipidemia
Dyslipidemias is one of the main risk factors for cardiovascular diseases, closely related to the metabolic syndrome and the condition of obesity [12].
Liver cells are primarily affected by ectopic accumulation of lipids as the liver is the major regulator of systemic accumulation of lipids and glucose. Fatty liver is associated with dyslipidemia and dysglycemia independently of visceral fat [13]. Consequently, NAFLD and NASH are the most common liver disorders in dyslipidemia, strongly associated with insulin resistance, increased risk of progression to liver cirrhosis, and possible onset of HCC [14].
Adipocytes play a crucial role in the tumor microenvironment through the secretion of several molecular mediators. In fact, adipose tissue secretes adipokines such as leptin, adiponectin, resistin, and inflammatory mediators, such as ANGPTL2, which modulate insulin sensitivity and trigger chronic low-grade inflammation. A dysregulated secretion of adipokines by adipocytes contributes to the development of obesity-related metabolic disorders [15].
The importance of dyslipidemia in the onset of NAFLD and the correlated HCC-NAFLD is explained by the suggestion of the use of statins as anti-inflammatory, anti-angiogenic, and anti-proliferative drugs [16]. These effects are not directly referable to the action of the drug but to the preventive action on lipotoxicity [14].
5. Smoke
Smoking has been associated with an increased risk for the development of HCC [17][18], although no studies have specifically examined the association between smoking and NAFLD-related HCC.
Tobacco carcinogens are metabolized in the liver, and the formation of DNA adducts could constitute the important initiator of hepatocarcinogenesis [19].
6. Gut Microbiota
Alterations of the intestinal microbiota, namely dysbiosis, have been associated with the spectrum of NAFLD [20][21]. Moreover, in fecal samples of cirrhotic patients with HCC, an overall decrease in microbial diversity with an increase in Gram-negative bacteria, predominantly Escherichia coli, has been reported [22][23]. The disruption of intestinal enterocyte intercellular tight junctions contributes to the onset of NAFLD, increasing gut permeability and translocation of gut bacteria (mainly Gram-negative bacteria) and lipopolysaccharides; this stimulates TLR4 at the hepatic level, leading to hepatic inflammation and fibrosis [22][24].
The gut microbiota are involved in choline metabolism, whose reduced levels are reflected in the liver, where they cause abnormal phospholipid synthesis and VLDL secretion. At least eight microbial species present in the intestine promote the metabolization of choline to TMA (trimethyllamine). From a clinical point of view, in addition to the hepatic consequences resulting from the very low secretion of VLDL, there is an increased risk of cardiovascular and renal diseases due to the hepatic metabolization of TMA into TMAO (trimethyllamine-N-oxide) [22].
With regard to NAFLD-HCC specifically, a recent work by Ponziani et al. demonstrated that those NAFLD subjects with HCC and cirrhosis have a peculiar gut microbiota profile with a lack of protective species compared to cirrhotic patients without HCC. This finding was associated with an enhanced intestinal inflammation that may have favored hepatocarcinogenesis through the expression of several inflammatory cytokines and chemokines, also opening the discussion on a possible therapeutical role of gut microbiota modulating agents (i.e., probiotics) or fecal transplantation in preventing HCC in NAFLD patients [25].
The intestinal microbiota also has a role in controlling the composition of bile acids [20][26]. Bile acids have a metabolic effect on NAFLD predominantly through two nuclear receptors: FXR (farnesoid X receptor) for primary bile acids and TGR5 for secondary bile acids. FXR activation is due to either bile acids themselves or FGF19, a gut hormone released in response to FXR activation. This pathway also affects glucose homeostasis and lipogenesis, reducing de novo synthesis and promoting β-oxidation of fatty acids, maintaining blood glucose and lipid levels in a normal range. TGR5 affects glucose homeostasis, energy expenditure by activation of thyroid hormones, and inflammation, which is negatively regulated [27][28].
Finally, patients with NAFLD have an alteration in the ratio of secondary to primary bile acids with loss of the beneficial antisteatotic and anti-inflammatory effects, and a higher concentrations of bile acids in the hepatic circulation. High levels of bile acids are able to activate inflammatory- and oxidative-stress-mediated cell death pathways, suggesting that bile acids may be involved in the pathogenesis of liver injury and potentially initiation of cancerous activity, particularly in the colon or the liver, where the secondary bile acids concentrate [29][30].
7. Genetics
Genetic factors are thought to contribute to 30–50% of diseases such as obesity, type 2 diabetes mellitus, atherosclerotic disease, and cirrhosis. Genetic polymorphisms (SNPs) in a number of genes have been associated with the presence of NAFLD and risk of disease progression to advanced fibrosis and HCC [31].
Two genes are considered most involved in the predisposition and development of NAFLD: patatin-like phospholipase domain-containing protein 3 (PNPLA3) and transmembrane 6 superfamily member 2 (TM6SF2).
The PNPLA3 mutation rs738409, encoding an I148M mutation, is independently associated with NAFLD, fibrosis progression, and an increased risk of HCC development [32]. This SNP has been reported to impair mobilization of triglycerides from hepatic lipid droplet, leading to an increase in hepatic fat content but not with alterations in glucose homeostasis and lipoprotein metabolism [33].
In a multivariate analysis that also included the presence of diabetes, BMI, age, and gender, the presence of the PNPLA3 mutation was shown to increase the risk of HCC by 2.3 times in heterozygotes and by 5 times in homozygotes [33].
The rs58542926 variant in the TM6SF2 gene, encoding an E167K mutation, is associated both with hepatic steatosis and an increased risk of liver fibrosis; however, its role in HCC development remains uncertain. The accumulation of triglycerides in the liver is due to the loss of function of this transporter with the inability to secrete lipoproteins rich in triglycerides and apolipoprotein. However, the inability to secrete VLDL reduces the incidence of cardiovascular disease in carriers of this polymorphism [34].
In a further study on individuals of European origin, the SNP rs641738 in the locus near the MBOAT7/TMC4 gene has been demonstrated to be associated with the severity of NAFLD [35]. This association is mediated by a decreased protein expression of MBOAT7 with consequent changes in the remodeling of the hepatic phosphatidylinositol acyl chain [36].
A study carried out in the UK found two mutations responsible for insulin resistance: the mutation (rs1044498, K121Q) of the ENPP1 gene and the mutation (rs1801278, Q972R) in the insulin receptor substrate-1 (IRS-1); both mutations, by reducing insulin sensitivity, were, independently of other factors, involved in NAFLD with a higher risk of progression to fibrosis [31].
Glucokinase regulatory protein (GCKR) regulates glucokinase activity and has been associated with NAFLD in the presence of the P446L mutation, which reduces the ability of GCKR to inhibit glucokinase in response to fructose-6-phosphate, thereby increasing the activity of the glucokinase and hepatic glucose absorption. The resulting uncontrolled hepatic glycolysis reduces glucose and insulin levels and increases the production of malonyl-CoA, promoting hepatic lipid accumulation. GCKR variants have been associated with fibrosis following NASH [37].
Considering the genes involved in oxidative stress, individuals carrying the variant SNP rs4880 of SOD2 have a 1.56-fold increased risk of developing advanced fibrosis [31].
An important role in the progression of fatty liver disease is also played by epigenetic regulation. Methylation of genes generally leads to a reduction in the expression of the gene product. Hypermethylation of the 99 CpG island in the regulatory region of PNPLA3 affects its expression and has been associated with advanced liver fibrosis. Furthermore, CpG99 methylation levels and PNPLA3 mRNA are affected by the PNPLA3 rs738409 genotype [38].
More recently, genetic variants have been combined into “polygenic risk scores”. These genetic variants are associated with HCC risk in individuals with multiple underlying liver diseases [39][40].
This entry is adapted from the peer-reviewed paper 10.3390/cancers15225458
References
- Kawamura, Y.; Arase, Y.; Ikeda, K.; Seko, Y.; Imai, N.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Sezaki, H.; Akuta, N.; et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am. J. Gastroenterol. 2012, 107, 253–261.
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of Metabolic Traits on the Risk of Cirrhosis and Hepatocellular Cancer in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 808–819.
- Hagström, H.; Tynelius, P.; Rasmussen, F. High BMI in late adolescence predicts future severe liver disease and hepatocellular carcinoma: A national, population-based cohort study in 1.2 million men. Gut 2018, 67, 1536–1542.
- Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Luo, J. Excess body weight and the risk of primary liver cancer: An updated meta-analysis of prospective studies. Eur. J. Cancer 2012, 48, 2137–2145.
- Yang, J.; He, J.; Feng, Y.; Xiang, M. Obesity contributes to hepatocellular carcinoma development via immunosuppressive microenvironment remodeling. Front. Immunol. 2023, 14, 1166440.
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Fedirko, V.; Jenab, M.; Trepo, E.; Boffetta, P.; Dahm, C.C.; Overvad, K.; Tjønneland, A.; et al. Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int. J. Cancer 2013, 132, 645–657.
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95.
- Raff, E.J.; Kakati, D.; Bloomer, J.R.; Shoreibah, M.; Rasheed, K.; Singal, A.K. Diabetes Mellitus Predicts Occurrence of Cirrhosis and Hepatocellular Cancer in Alcoholic Liver and Non-alcoholic Fatty Liver Diseases. J. Clin. Transl. Hepatol. 2015, 3, 9–16.
- Yang, J.D.; Ahmed, F.; Mara, K.C.; Addissie, B.D.; Allen, A.M.; Gores, G.J.; Roberts, L.R. Diabetes Is Associated with Increased Risk of Hepatocellular Carcinoma in Patients with Cirrhosis from Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 907–916.
- Nilsson, P.M.; Tuomilehto, J.; Ryden, L. The metabolic syndrome—What is it and how should it be managed? Eur. J. Prev. Cardiol. 2019, 26, 33–46.
- Speliotes, E.K.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Meigs, J.B.; Sahani, D.V.; Hirschhorn, J.N.; O’Donnell, C.J.; Fox, C.S. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The Framingham Heart Study. Hepatology 2010, 51, 1979–1987.
- Rajesh, Y.; Sarkar, D. Association of Adipose Tissue and Adipokines with Development of Obesity-Induced Liver Cancer. Int. J. Mol. Sci. 2021, 22, 2163.
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997.
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238.
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e417.
- Abdel-Rahman, O.; Helbling, D.; Schöb, O.; Eltobgy, M.; Mohamed, H.; Schmidt, J.; Giryes, A.; Mehrabi, A.; Iype, S.; John, H.; et al. Cigarette smoking as a risk factor for the development of and mortality from hepatocellular carcinoma: An updated systematic review of 81 epidemiological studies. J. Evid. Based Med. 2017, 10, 245–254.
- Petrick, J.L.; Campbell, P.T.; Koshiol, J.; Thistle, J.E.; Andreotti, G.; Beane-Freeman, L.E.; Buring, J.E.; Chan, A.T.; Chong, D.Q.; Doody, M.M.; et al. Tobacco, alcohol use and risk of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: The Liver Cancer Pooling Project. Br. J. Cancer 2018, 118, 1005–1012.
- Mouzaki, M.; Loomba, R. Insights into the evolving role of the gut microbiome in nonalcoholic fatty liver disease: Rationale and prospects for therapeutic intervention. Ther. Adv. Gastroenterol. 2019, 12, 1756284819858470.
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775.
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease: From Composition to Function. Clin. Gastroenterol. Hepatol. 2019, 17, 296–306.
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Masior, L.; Lewandowski, Z.; Kosinska, I.; Grat, K.; Stypulkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated with the Presence of Hepatocellular Cancer in Patients with Liver Cirrhosis. Transpl. Proc. 2016, 48, 1687–1691.
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887.
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated with Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120.
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e1673.
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573.
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539.
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328.
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128.
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744.e1727.
- Stender, S.; Loomba, R. PNPLA3 Genotype and Risk of Liver and All-Cause Mortality. Hepatology 2020, 71, 777–779.
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465.
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514.
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e1216.
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265.
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088.
- Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Imajo, K.; Saito, S.; Yoneda, M.; Nakamura, T.; Nakajima, A.; Hotta, K. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 494–502.
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782.
- Gellert-Kristensen, H.; Richardson, T.G.; Davey Smith, G.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Stender, S. Combined Effect of PNPLA3, TM6SF2, and HSD17B13 Variants on Risk of Cirrhosis and Hepatocellular Carcinoma in the General Population. Hepatology 2020, 72, 845–856.
This entry is offline, you can click here to edit this entry!