Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Alkaloids found in multiple species, known as ‘driver species’, are more likely to be included in early-stage drug development due to their high biodiversity compared to rare alkaloids. Many synthetic approaches have been employed to hybridize the natural alkaloids in drug development. Click chemistry is a highly efficient and versatile reaction targeting specific areas, making it a valuable tool for creating complex natural products and diverse molecular structures. It has been used to create hybrid alkaloids that address their limitations and serve as potential drugs that mimic natural products.
- heterocycles
- 1,2,3-triazole
- alkaloids
- biological properties
- molecular hybridization
1. Introduction
1.1. Natural Products as a Source of Bioactive Compounds
Natural products (NPs) are a valuable source of therapeutic substances due to their intricate and varied structures [1][2][3]. NPs have been instrumental in drug discovery, especially in treating cancer and infectious diseases [4][5][6]. Their diverse stereo centers, sp3 carbon, and labile functionalities have been instrumental. NPs have also been effective in treating other medical conditions such as cardiovascular diseases (such as statins) and multiple sclerosis (such as fingolimod) [7][8][9]. Their larger molecular mass, higher number of sp3 carbon and oxygen atoms, and lower number of nitrogen and halogen atoms contribute to their unique molecular scaffolds, offering a wealth of structural diversity for drug development [10][11]. Over 60% of the drugs on the market are derived from NPs, highlighting their importance in the pharmaceutical industry [12]. However, the complex nature of NPs often requires complicated synthetic strategies and time-consuming multistep syntheses, resulting in limited yields and derivatives. Therefore, simple, modular, selective, and reliable chemistry is necessary for the late-stage functionalization and diversification of natural products. Alkaloids are an essential class of compounds found in natural products.
Alkaloids are small organic molecules that contain nitrogen, usually in a ring, and are present in plants (about 20% of plant species) [13][14][15]. They have a noticeable impact on the physiology of both humans and animals. Common examples of alkaloids used as drugs include atropine, berberine, cocaine, codeine, capsaicin, ephedrine, morphine, nicotine, quinine, papaverine, reserpine, strychnine, vinblastine, vincristine, etc., and are known for their diverse biological properties [16][17][18][19]. Modifying the chemical structure of known alkaloids is an essential component of drug discovery, as it enables researchers to study the relationship between structure and activity and uncover new biological responses [20].
Drug discovery in modern times involves molecular hybridization, which is used for rationally designing drugs [21][22][23]. This method combines different pharmacophoric moieties of bioactive substances covalently, creating new hybrid compounds that can be more effective than their parent pharmacophores. Hybridizing various organic molecular motifs with different pharmacological activities can create compounds with modified selectivity profiles, different modes of action, reduced side effects, the ability to overcome multidrug resistance, and an improved safety profile [24][25][26][27][28][29][30][31]. This approach helps with the design of multifunctional drug candidates and can overcome the limitations associated with current drugs by providing various advantages (see Figure 1).
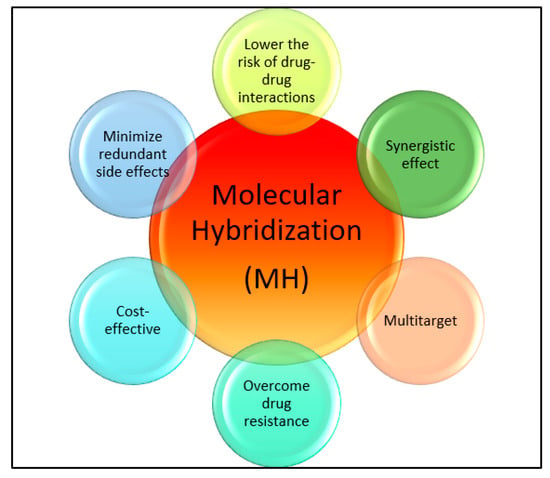
Figure 1. Importance of molecular hybridization.
1.2. Click Chemistry: Principles and Applications
In 2001, Sharpless and his team introduced the concept of click chemistry, which is now widely used in the scientific community. Click chemistry is a simple and efficient way to join two molecular building blocks under mild and adaptable conditions, resulting in high yields while requiring minimal purification. This method is user-friendly and can be compared to fastening two seatbelt ends together. The versatility of click chemistry has made it an indispensable tool in various fields such as analytical chemistry, materials science, chemical biology, and drug development [32][33][34].
Click chemistry involves merging small molecular units to form new molecular attributes. Nevertheless, individuals in the scientific and engineering fields, who need to possess the proper skills and equipment to execute such connecting operations with consistency, can face difficulties in this domain. Fortunately, the chemical strategies available today, among them click chemistry, simplify the process of molecular connectivity with innovative methods continuously emerging [35].
A “click reaction” is simply a reaction forming new carbon–heteroatom bonds in high yields and selectivity. The term “click” was later coined to indicate the ease of execution, and is synonymous with buckling (clicking) a seatbelt. The most widely used click reaction in medicinal chemistry and biochemistry is the formation of a 1,2,3-triazole moiety by combination of an azide and alkyne. Specific examples that form the 1,2,3-triazole functionality include copper-catalyzed azide–alkyne cycloaddition (CuAAC), strain-promoted azide–alkyne cycloaddition (SPAAC), photoinduced click reactions (light-triggered), and bioorthogonal click chemistry reactions [36][37][38][39][40][41][42]. These reactions are defined as 1,3-dipolar cycloadditions between an alkyne and azide functional group, resulting in a 1,4-disubstituted 1,2,3-triazole as a linker between the two coupled units (Figure 2).
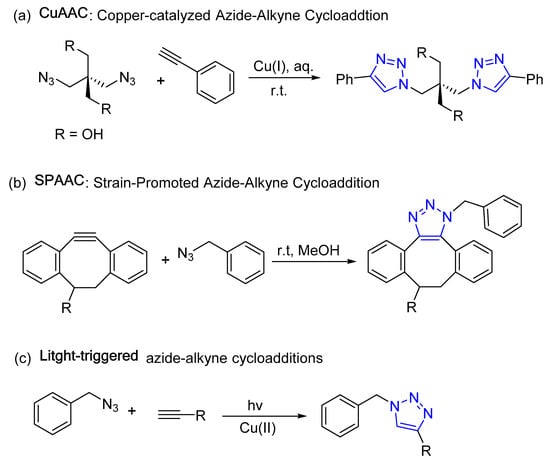
Figure 2. Representative 1,2,3-triazole-based click chemistry reactions.
In contrast to the classic thermal (Huisgen) 1,3-dipolar reactions, click chemistry products are generated with high regioselectivity, eliminating the possibility of regioisomers [43][44]. These reactions occur quickly and are thermodynamically driven, as the formation of triazoles is highly exothermic. Click chemistry is also flexible and can tolerate various functional groups, making it easy to synthesize complex molecules. This is helpful in various industries, such as enhancing natural products, polymers, biomolecules, and macrocyclic structures by attaching molecular tags precisely and efficiently [45]. The 1,2,3-triazole scaffold has proven to play a crucial role in the biological properties of various bioactive molecules, showcased in Figure 3, with several FDA-approved drugs containing this scaffold.
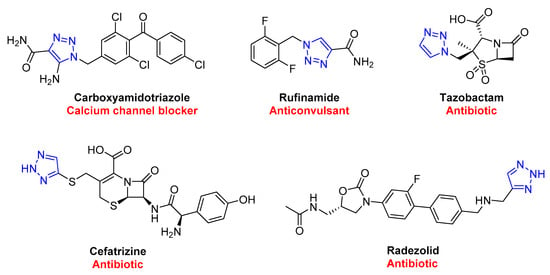
Figure 3. FDA-approved drugs containing 1,2,3-triazole scaffold.
2. Application of Click Chemistry with Natural Product Hybridization
The integration of click chemistry with natural product modification has opened new avenues for exploring the therapeutic potential of natural products. By attaching molecular tags, functional groups, or probes to natural products through click chemistry, researchers can enhance their properties, improve their stability, facilitate their characterization, and enhance selectivity. The tagged natural products can be utilized in various applications, including drug discovery, target identification, chemical biology, proteomics, and imaging [46][47][48].
Molecular Hybridization of Alkaloids Using Click Chemistry
Alkaloids are a diverse group of nitrogen-containing secondary metabolites in plants, animals, marine, and microbes [49][50][51][52]. These compounds are typically formed from amino acids and can have a variety of chemical structures. Many alkaloids have been isolated from plants, and have been used to create valuable drugs in modern medicine [53][54][55]. Alkaloids have been shown to have therapeutic properties such as anesthetics, cardioprotective agents, and anti-inflammatory agents. Some well-known alkaloids commonly used in clinical settings include morphine, strychnine, quinine, ephedrine, and nicotine. Recently, there has been a renewed interest in bioactive natural products due to the potential for drug discovery and the development of traditional medicines. Various organic chemistry strategies have extensively explored the click chemistry approach to develop potential drug candidates.
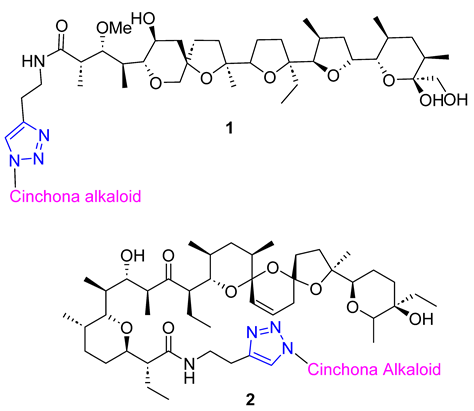
Skiera et al. used cinchona azides and homologated azides to create new 1,2,3-triazole hybrids in combination with antibiotic polyether ionophores, specifically salinomycin and monensin N-propargylamides 1 and 2. Despite their complex structure, the standard CuAAC protocol was highly efficient in producing the desired products. In vitro testing against three cancer cell lines (LoVo, LoVo/DX, and HepG2) revealed that two compounds had significant antiproliferative activity, with IC50 values below 3.00 μM. Furthermore, these compounds were more selective towards normal cells than commonly used anticancer drugs [56].
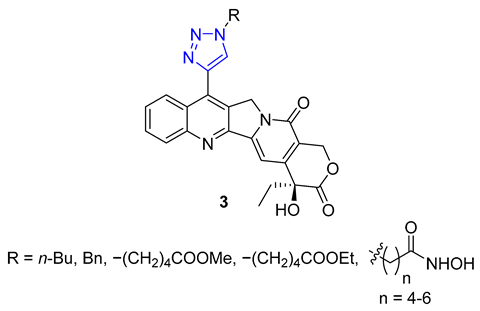
Huang and colleagues successfully created a group of 7-triazole-substituted camptothecin 3 through the CuAAC reaction. The process started with 7-ethynylcamptothecin, which combined 7-chlorocamptothecin with Sonogashira coupling. The alkyne was then combined with different azides using the standard CuAAC protocol, resulting in the corresponding triazoles with high yields. After removing the camptothecin ester, the final products 3 were tested for cytotoxicity against several cell lines, including A549, HCT-116, HT-29, LoVo, and MDA-MB-231. Three products with R = n-Bu, Bn, and −(CH2)4COOMe showed excellent in vitro activity. Moreover, all of the products 3 maintained their inhibitory ability against Topoisomerase I [57].
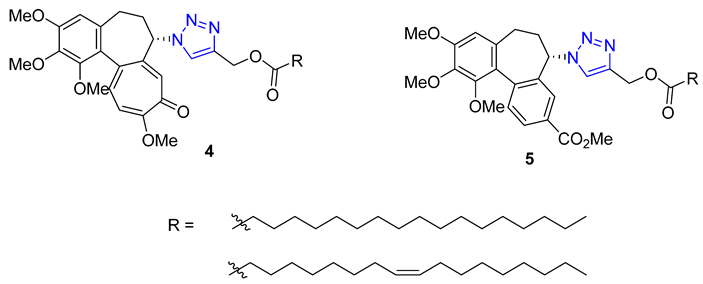
Kuznetsova and Schmalz envisioned making colchicine less toxic and improving its distribution in the body by employing lipophilic groups to the molecule. They used a two-step process that involved reacting colchicine and allocolchicine with propargyl alcohol and then esterifying them with palmitic or oleic acids to create lipophilic triazoles. During testing, they found that only the colchicine derivatives 4 were effective, while the allocolchicine derivatives 5 were less effective. One palmitic ester showed a significant increase in antimitotic activity, but had a lower affinity to the colchicine binding site in tubulin. They then tested these esters by encapsulating them in liposomes made with egg phosphatidylcholine, yeast phosphatidylinositol, and palmitic or oleic esters. The liposomal formulation of the ester with the oleoyl chain was more effective in inhibiting cell proliferation than the unencapsulated ester. Screening these liposomes against four human tumor cell lines confirmed the retained cytotoxicity of colchicine derivatives [58][59].
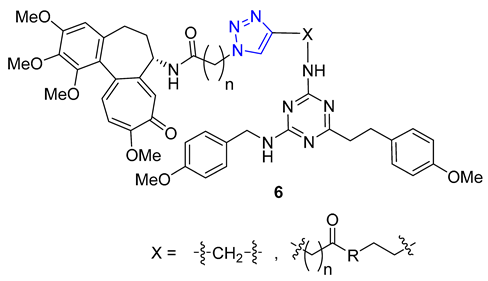
A group of researchers, led by Fedorov, created a set of two-agent combinations called dyads, consisting of colchicine and tubulizine, which are both antimitotic agents. The purpose of this was to enhance their individual anticancer abilities. The process involved using the CuAAC reaction of azide colchicine congeners with acetylene-decorated tubulizine, resulting in products 6 with high yields. All dyads portrayed significant cytotoxic activity against the HBL100 human mammary cell line, with IC50 values varying from 0.60 to 2.93 μM. Although all triazoles showed greater activity than tubulizine, none were more effective than deacetylcolchicine. Furthermore, some compounds in this series acted as sub-stoichiometric inhibitors of microtubule assembly [60].
Shi and co-workers utilized berberine as a scaffold to synthesize a novel class of triazole-containing derivatives through click chemistry. The primary objective of this research was to develop potent multivalent inhibitors targeting acetyl- and butyrylcholine esterases (AChE and BChE), as well as to explore their potential as β-amyloid aggregation inhibitors, which play a crucial role in the neurodegenerative cascade of Alzheimer’s disease. The synthetic pathway involved converting berberine azides into partially demethylated berberrubine. Subsequently, N-propargyl-substituted tertiary amines were obtained by reacting secondary amines with propargyl bromide. The CuAAC reaction successfully yielded the desired berberine triazoles. Screening of the synthesized compound library led to the identification of several highly active compounds. Compound X (R = 4-diisopropylamine) demonstrated remarkable inhibitory activity against AChE, with an impressive IC50 value of 0.044 μM, displaying selectivity towards AChE. Additionally, when the triazole ring bore a dibutylamine substituent, the conjugate exhibited good inhibitory activity against AChE, with an IC50 value of 0.20 μM, and displayed the highest potency in inhibiting β-amyloid aggregation [61].
Jin and Yan extended the previous study, focusing on preparing a library of berberine triazoles with reverse connectivity of the triazole group to evaluate their cytotoxicity. O-propargylation of berberrubine was achieved directly, yielding O-propargylberberine, which was subsequently subjected to a CuAAC reaction with substituted benzyl azides to produce the corresponding triazoles. All synthesized products were evaluated against three human cancer cell lines (MCF-7, SW-1990, and SMMC-7721) and the HUVEC line (normal human umbilical vein endothelial cell) to assess their potential as anticancer agents. Remarkably, most of the derivatives displayed significantly higher anticancer activities against MCF-7 cells than the parent berberine. Compound 8, featuring a 4-tert-butyl substituent in the phenyl ring, exhibited the highest potency against the SW-1990 and SMMC-7721 cell lines, with IC50 values of 8.5 and 11.9 μM, respectively [62].
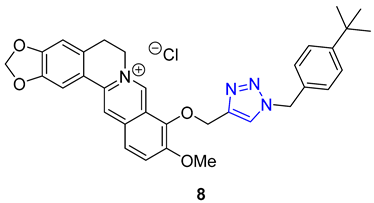
Pingaew et al. synthesized a novel 1,2,3,4-tetrahydroisoquinoline derivative containing a 1,2,3-triazole moiety using a modified Pictet–Spengler reaction and the CuAAC method to introduce the triazole ring. The antiproliferative activity of the products was evaluated against four cancer cell lines (HuCCA-1, HepG2, A549, and MOLT-3), and some compounds (9) displayed significant cytotoxicity. Notably, compounds with R = o-aryl methyl ester demonstrated exceptional potency against HuCCA-1 (IC50 = 0.63 μM) and A549 (IC50 = 0.57 μM) cell lines. Additionally, triazole X (R = p-Toluenyl) exhibited remarkable activity against HepG2 cells (IC50 = 0.56 μM), and surpassed the activity of drugs such as etoposide or doxorubicin without harming normal cells [63].
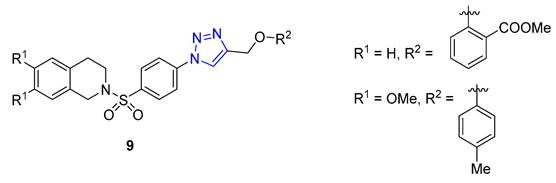
Researchers led by Melander tackled the challenge of fighting biofilm-forming bacteria, which are responsible for 75% of human body infections. They turned to the flustramine family of alkaloids to combat this issue, known for their broad antimicrobial activity. The researchers used them as a scaffold for synthesizing an 18-member library of pyrroloindoline 1,2,3-triazole amides through CuAAC synthesis. The team created the library by reacting various azides with a synthetic alkyne-functionalized pyrroloindoline mimic of flustramine, then deprotecting the pyrrolidine nitrogen atom. The library was then screened against Gram-positive and Gram-negative bacterial strains, including A. baumannii, E. coli, and MRSA (methicillin-resistant Staphylococcus aureus), to determine its ability to modulate biofilm formation. The screening resulted in the identification of several nontoxic compounds with low micromolar IC50 values. Compound 10 showed high activity against MRSA with p-alkylphenyl groups (C5–C7). It effectively inhibited biofilm formation in methicillin-sensitive strain Staphylococcus aureus with IC50 values ranging from 6.6 to 32.0 μM [64].
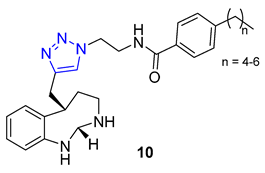
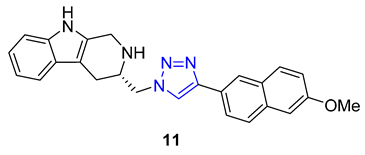
Ochrolifuanine E, a bis-indole alkaloid found in the Thai herbal medicine plant Dyera costulata, emerged as a potential inhibitor of β-secretase (BACE1) through a virtual screening procedure conducted by Vajragupta’s group. BACE1 plays a critical role in producing amyloid peptides from the amyloid precursor protein, making it a promising target for Alzheimer’s disease drug development. Docking studies of the parent alkaloid revealed that tryptoline (2,3,4,9-tetrahydro-1H-pyrido [3,4-b]indole) served as the key pharmacophore responsible for the enzyme binding. This insight led to the design of a library of 1,2,3-triazolyl tryptoline derivatives from which 22 most promising candidates, selected based on docking analysis, were synthesized using the CuAAC reaction. Screening of these derivatives for inhibitory activity against BACE1 resulted in the identification of a potent inhibitor, compound 11 (IC50 = 1.49 μM), exhibiting 100 times greater selectivity to BACE1 over Catepsin D [65].
Shinde and colleagues presented a new approach to creating 1,2,3-triazole-substituted piperidines. They achieved this by adding an azido group to the fourth position of the piperidine ring, using a simple transformation of N-Boc-protected piperidinone. The resulting azide was then combined with either ethyl propiolate or propargyl alcohol, using CuI as a catalyst to produce the desired products. These products were then further modified to create derivatives 12 and 13, exhibiting exceptional antifungal properties [66][67].
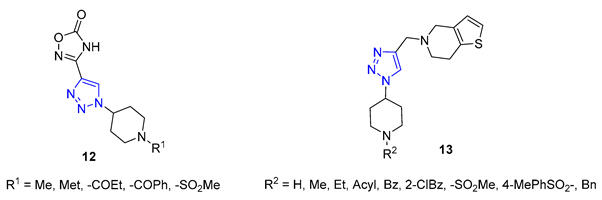
Da Silva utilized 6-mercaptopurine, a synthetic drug that is similar to naturally occurring purine alkaloids, to create a new series of mono- and bis-1,2,3-triazolyl derivatives. He first conducted N- and S-propargylation of 6-mercaptopurine to produce mono- or dipropargyl derivatives. These were then subjected to the CuAAC reaction with azidoacetic acid and methyl 3-β-azidocholanoate. From a range of products, compounds 14 and 15 demonstrated exceptional antimalarial activity in vitro, with a higher rate of parasite multiplication inhibition compared to the common antimalarial drug, chloroquine [68].
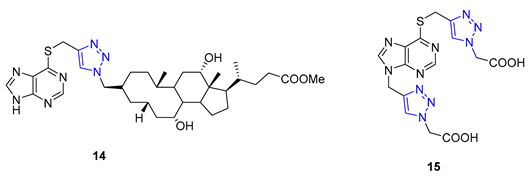
In a recent study, Nair and colleagues explored the potential of purine as a building block for developing novel neuroprotective agents. Their approach involved integrating a 1,2,3-triazole ring and fluoroaromatic groups into the purine scaffold. To begin, the researchers modified commercially available 2,6-dichloropurine by introducing an alkyne group through a two-step process. They then utilized the CuAAC reaction within situ-generated benzyl azides to construct a series of compounds, including Compound 16. These compounds were evaluated for their ability to safeguard neurons using fluorescence electron microscopy. Interestingly, one of the derivatives containing an o-fluorophenylmethyl group exhibited neuroprotective properties on par with flavopiridol and roscovitine, two well-known cyclin-dependent kinase (CDK) inhibitors. This compound effectively mitigated amyloid β (Aβ)-induced neurotoxicity [69].
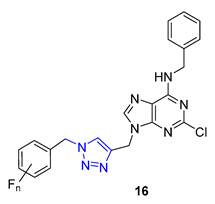
The Wang group developed glycoconjugates of phenanthroindolizidine alkaloids that target tobacco mosaic virus (TMV). The team utilized three different methods to conjugate (S)-6-O-desmethylantofine and 14-hydroxytylophorine with sugars. One of the methods involved creating a 1,2,3-triazole linker to attach O-propargylated alkaloids with three different monosugar units. The resulting glycoconjugates, 17 and 18, demonstrated improved water solubility. However, their activity was only moderate compared to glycoconjugates linked by simple glycosidic bonds, which showed highly active results. These findings highlight the significance of the glycosidic bond in creating potent activity in glycoconjugates that target TMV [70].
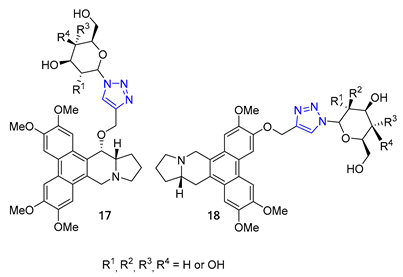
Carroll and their team have effectively created a range of cocaine-like compounds called 2β-alkynyl and 2β-(1,2,3-triazolyl)-3β-(aryl)tropanes. They tested the compounds’ ability to bind to dopamine, serotonin, and norepinephrine membrane transporters using radioligand binding assays. Surprisingly, all of the substances produced from the alkyne intermediate through the CuAAC reaction, including two triazoles designated as 19, demonstrated a strong affinity for the dopamine transporter (DAT) in nanomolar or sub-nanomolar concentrations [71].
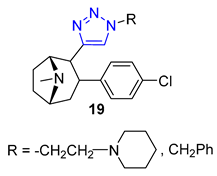
Researchers Thuy et al. have investigated the properties of Murrayafoline A, a carbazole alkaloid found in the roots of different Murraya species. This substance has a wide range of biological activities, including the ability to kill fungi and induce apoptosis. To explore its potential further, the researchers synthesized a series of derivatives called X by combining the azido derivative of Murrayafoline A with various alkynes using a CuAAC reaction. They then evaluated the biological effects of these derivatives, focusing on their anti-inflammatory properties. The results showed that two of the triazoles 20 (R = −CH2NH2 and −CH2OH) were particularly effective at inhibiting the production of cytokines IL-12 p40, IL-6, and TNF-α, outperforming the unmodified Murrayafoline A [72].
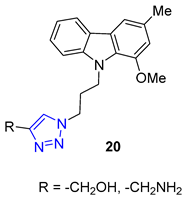
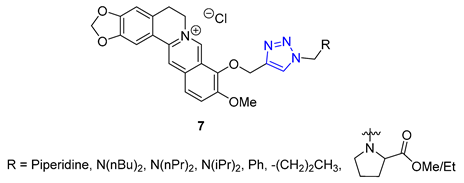
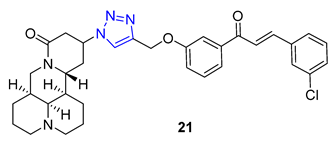
Matrine, a quinolizidine alkaloid derived from the root of Sophora flavescens Ait (also known as Kushen), is a traditional Chinese herb used for centuries for the treatment of liver disease. Zhao et al. focused on synthesizing matrine–triazole–chalcone hybrids and reported their notable growth inhibitory effects on a range of cancer cells. Compound 21 (IC50 = 5.01–7.31 μM) exhibited broad-spectrum anticancer activities against various cancer cell lines (A549, Bel-7402, Hela, and MCF-7). Notably, Compound 21 displayed superior potency compared to the combination of matrine and chalcone (IC50 > 50 μM) as well as 5-fluorouracil (IC50 = 8.93–40.38 μM). Structure–activity relationship (SAR) studies suggested that the α, β-unsaturated ketone moiety and the triazole played crucial roles in determining the enhanced inhibitory activity. Further investigations revealed compound 21 could induce apoptosis in A549 cells and effectively suppress tumor growth in an A549-xenografted nude mouse model (at 10 mg/kg) without causing apparent cytotoxicity [73].
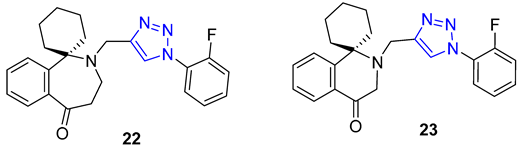
Homoharringtonine and homoerythrina, naturally occurring alkaloids isolated from the genus Cephalotaxus, have gained significant attention due to their derivatives’ diverse biological activities, particularly their potential as anticancer agents. In a study conducted by Li et al., homoerythrina–triazole 22 (IC50 = 1.89–4.19 μM) displayed more potent inhibitory activity than rucaparib (IC50 = 4.91–13.51 μM) and harringtonine (IC50: 10.55–11.71 μM) in A549, HCT-116, and MCF-7 cancer cells. Mechanistic investigations revealed that 22 arrested the cell cycle at the S phase, inhibited PAR biosynthesis, and induced apoptosis in A549 cells. Additionally, the same research group reported erythrina–triazole 23 (IC50 = 0.23–1.13 μM) to exhibit superior inhibitory activity compared to rucaparib (IC50 = 2.58–13.82 μM) across a panel of cancer cell lines (A549, OVCAR-3, HepG2, A375, and SW-620). Mechanistic studies indicated Compound 23 inhibited PAR biosynthesis and induced apoptosis in A549 cells [74][75].
Combining two different natural products via the CuAAC reaction has proven to be an effective method for creating new, functional compounds. One such compound is cytisine–triazole–camphor 24, reported by Artyushin et al., which was found to have strong antiviral properties against influenza virus A/Puerto Rico/8/34 (H1N1), with low toxicity and an impressive selectivity index (IC50 = 8 μM, CC50 = 168 μM, SI = 20). Notably, its selectivity index is higher than that of the reference drug rimantadine (IC50 = 67 μM, CC50 = 335 μM, SI = 5) [76].
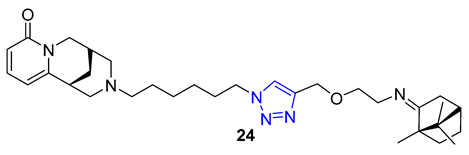
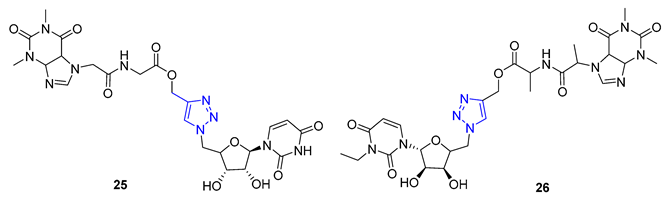
Theophylline, a naturally occurring purine base, serves as a bronchodilator drug for the treatment of respiratory diseases such as chronic pulmonary obstructive disease and asthma. Ruddarraju et al. sought to explore its antibacterial properties in vitro by linking theophylline and nucleosides by incorporating a triazole ring using conventional Huisgen conditions. The triazole-tethered theophylline–nucleoside hybrid 25 exhibited significant growth inhibition against A549, HT-29, MCF-7, and A375 cancer cells (IC50 = 1.89–4.89 μM). On the other hand, hybrid 26 displayed potent antibacterial activities against both Gram-positive strains (Staphylococcus aureus, Bacillus cereus) and Gram-negative strains (Escherichia coli and Pseudomonas aeruginosa) with impressive MIC values (MIC = 0.03125–0.125 μg/mL), comparable to or even more potent than the clinical drug ciprofloxacin (MIC = 0.0156–0.0625 μg/mL) [77][78].
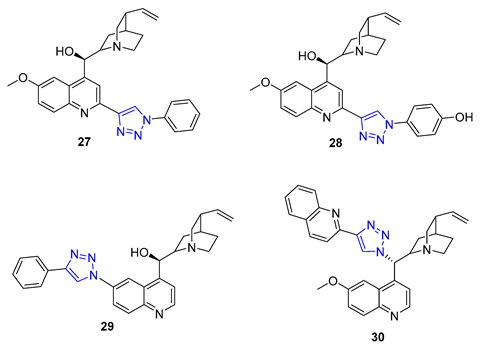
Quinine, a natural cinchona alkaloid, is a crucial element in antimalarial drugs and is widely accessible. Boratyński et al. recently created a range of quinine derivatives by introducing azide at C9, C2’, and C6’. This resulted in a focused library of triazole-containing chinchona alkaloids (27–30). In vitro studies revealed that most of these derivatives possessed moderate antiproliferative activity. Among them, 27 (IC50 = 0.53 μM) displayed the most potential in MC-4-11 cells, while 28 (IC50 = 1.2 μM) was most effective in HT-29 cells [79].
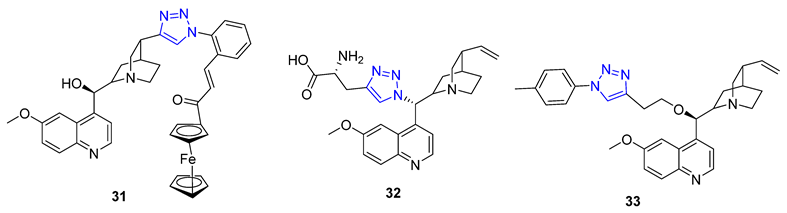
New molecules can have unexpected properties when small molecules are combined with ferrocene, a unit known for its adjustable redox characteristics. Pešić et al. discovered that 31 with IC50 value 2.34–2.13 μM, a ferrocene–quinine conjugate, inhibited the growth of both drug-sensitive NCI-H460 cancer cells and multi-drug resistant (MDR) NCI-H460/R cancer cells. Mechanistic studies showed that 31 increased ROS production and induced mitochondrial damage in MDR cancer cells, highlighting the importance of the ferrocene compound [80]. Meanwhile, Sahu et al. reported that 32, a C19 quinine–triazole derivative, had potent antimalarial with IC50 = 0.25 μM against P. falciparum and antileishmanial activities (L. donavani, IC50 = 1.78 μM) with no apparent adverse effects. Structural toxicological activity relationship studies suggested that including the triazole compound in quinine decreased toxicity [80]. Additionally, Panda et al. found that 33 exhibited more potent in vitro antimalarial activity with an IC50 value of 27 nM than quinine (IC50 = 58 nM) against P. falciparum strain 3D7, likely due to the addition of a hydrophobic alkyl chain at C9 that improved the scaffold’s penetration ability [81].
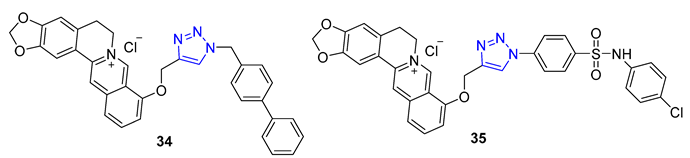
Berberine is a type of isoquinoline alkaloid that can be found in different Berberis plants. Berberine–triazoles have been found to possess both anticancer and antimalarial properties. In a study conducted by Sun et al., it was discovered that a compound called 34 could inhibit the growth of SW-1990 (IC50 = 22.2 μM) and SMMC-7721 (IC50 = 14.9 μM) cancer cells [82]. Meanwhile, Nath et al. reported that berberine–triazole 35 (IC50 = 0.142 μM) could exert antimalarial activity against the P. falciparum (3D7) strain without causing any apparent harm to human PC-3 cells (IC50 > 200 μg/mL) [83].
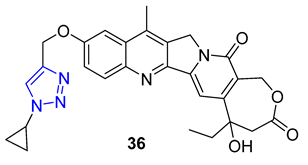
In 1966, Camptotheca acuminata yielded 20(S)-Camptothecin, a powerful inhibitor of DNA topoisomerase I. Xu and colleagues later synthesized a range of C10 homocamptothecin–triazole derivatives by introducing an alkyne at C10 of homocamptothecin and conducting reactions with various azides under CuAAC. Among them, derivative 36 (IC50 = 30 nM) was found to be more effective at inhibiting A549 cancer cells in a Topo I-dependent manner than 20(S)-camptothecin (IC50 = 170 nM). According to mechanistic studies, 35 can halt the cell cycle at G2 and S phases [84].
3. Drug-like Properties
Alkaloids, like vinblastine, were originally discovered in plants and have been approved for cancer treatment. However, their effectiveness is limited by poor solubility, low bioavailability, drug resistance, and possible liver toxicity [85][86][87]. This has hindered further experimentation beyond lab testing. Therefore, it is crucial to thoroughly understand the drug-like properties of alkaloids when developing potential drug candidates and using them as scaffolds. Preclinical drug development involves ADMET studies, which stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity studies. These studies evaluate a drug candidate’s pharmacokinetic and pharmacodynamic properties. ADMET studies provide essential information about a drug’s efficacy, safety, and pharmacokinetic properties, which helps make informed decisions regarding dosage, formulation, and potential risks associated with the drug candidate. Integrating ADMET studies into natural product drug development provides researchers with valuable insights into these compounds’ pharmacokinetic and pharmacodynamic properties, which helps demonstrate their drug-like properties [88][89][90][91][92][93][94].
A molecule’s drugability can be estimated using Lipinski’s rule of five (RO5), determining if a biologically active chemical is bioavailable [95][96]. The rule states that certain molecular qualities, such as having no more than five hydrogen bond donors, a maximum of ten hydrogen bond acceptors, a mass of less than 500 Da, and a partition coefficient (logP) value of less than five, are linked to pharmacokinetic drug features such as absorption, distribution, metabolism, and excretion. If one or more of these conditions are violated, the molecule is predicted to be incapable of being taken orally. The number of rotatable bonds and polar surface areas are also significant factors in assessing oral bioavailability [97]. For example, the maximum number of rotatable bonds should be ten, and a molecule’s polar surface area should be no larger than 140 Å for orally active medicines carried by the transcellular route [98][99]. The hERG IC50 value is another critical characteristic determining a drug’s effectiveness in blocking potassium hERG channels.
This entry is adapted from the peer-reviewed paper 10.3390/molecules28227593
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; The international natural product sciences taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216.
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803.
- Sun, X.; Zhang, Y.; Zhou, Y.; Lian, X.; Yan, L.; Pan, T.; Jin, T.; Xie, H.; Liang, Z.; Qiu, W.; et al. NPCDR: Natural product-based drug combination and its disease-specific molecular regulation. Nucleic Acids Res. 2022, 50, D1324–D1333.
- Atanasov, A.G.; Waltenberger, , B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614.
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129.
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381.
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661.
- Waltenberger, B.; Mocan, A.; Šmejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural products to counteract the epidemic of cardiovascular and metabolic disorders. Molecules 2016, 21, 807.
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis—Success from bench to bedside. Nat. Rev. Neurol. 2019, 15, 53–58.
- Li, J.W.H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165.
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837.
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599.
- Srivastava, S.; Srivastava, A.K. Biotechnology and Genetic Engineering for Alkaloid Production. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 213–250.
- Bhambhani, S.; Kondhare, K.R.; Giri, A.P. Diversity in Chemical Structures and Biological Properties of Plant Alkaloids. Molecules 2021, 26, 3374.
- Srinivasan, P.; Smolke, C.D. Biosynthesis of medicinal tropane alkaloids in yeast. Nature 2020, 585, 614–619.
- Daley, S.K.; Cordell, G.A. Alkaloids in contemporary drug discovery to meet global disease needs. Molecules 2021, 26, 3800.
- Qiu, S.; Sun, H.; Zhang, A.; Xu, H.; Yan, G.; Han, Y.; Wang, X. Natural alkaloids: Basic aspects, biological roles, and future perspectives. Chin. J. Nat. Med. 2014, 12, 401–406.
- Heinrich, M.; Mah, J.; Amirkia, V. Alkaloids used as medicines: Structural phytochemistry meets biodiversity—An update and forward look. Molecules 2021, 26, 1836.
- Olofinsan, K.; Abrahamse, H.; George, B.P. Therapeutic Role of Alkaloids and Alkaloid Derivatives in Cancer Management. Molecules 2023, 28, 5578.
- Zhang, X.; Zhang, S.; Zhao, S.; Wang, X.; Liu, B.; Xu, H. Click Chemistry in Natural Product Modification. Front. Chem. 2021, 9, 774977.
- Ivasiv, V.; Albertini, C.; Goncalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular hybridization as a tool for designing multitarget drug candidates for complex diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711.
- Harrison, J.R.; Brand, S.; Smith, V.; Robinson, D.A.; Thompson, S.; Smith, A.; Davies, K.; Mok, N.; Torrie, L.S.; Collie, I.; et al. A molecular hybridization approach for the design of potent, highly selective, and brain-penetrant n-myristoyltransferase inhibitors. J. Med. Chem. 2018, 61, 8374–8389.
- Sampath Kumar, H.M.; Herrmann, L.; Tsogoeva, S.B. Structural hybridization as a facile approach to new drug candidates. Bioorg. Med. Chem. Lett. 2020, 30, 127514.
- Bokhtia, R.M.; Girgis, A.S.; Ibrahim, T.S.; Rasslan, F.; Nossier, E.S.; Barghash, R.F.; Sakhuja, R.; Abdel-Aal, E.H.; Panda, S.S.; Al-Mahmoudy, A.M.M. Synthesis, antibacterial evaluation, and computational studies of a diverse set of linezolid conjugates. Pharmaceuticals 2022, 15, 191.
- Panda, S.S.; Tran, Q.L.; Rajpurohit, P.; Pillai, G.G.; Thomas, S.J.; Bridges, A.E.; Capito, J.E.; Thangaraju, M.; Lokeshwar, B.L. Design, synthesis, and molecular docking studies of curcumin hybrid conjugates as potential therapeutics for breast cancer. Pharmaceuticals 2022, 15, 451.
- Panda, S.S.; Thangaraju, M.; Lokeshwar, B.L. Ursolic acid analogs as potential therapeutics for cancer. Molecules 2022, 27, 8981.
- Flint, A.L.; Hansen, D.W.; Brown, L.D.; Stewart, L.E.; Ortiz, E.; Panda, S.S. Modified curcumins as potential drug candidates for breast cancer: An overview. Molecules 2022, 27, 8891.
- Bokhtia, R.M.; Panda, S.S.; Girgis, A.S.; Samir, N.; Said, M.F.; Abdelnaser, A.; Nasr, S.; Bekheit, M.S.; Dawood, A.S.; Sharma, H.; et al. New NSAID conjugates as potent and selective COX-2 inhibitors: Synthesis, molecular modeling and biological investigation. Molecules 2023, 28, 1945.
- Wyman, K.A.; Girgis, A.S.; Surapaneni, P.S.; Moore, J.M.; Abo Shama, N.M.; Mahmoud, S.H.; Mostafa, A.; Barghash, R.F.; Juan, Z.; Dobaria, R.D.; et al. Synthesis of potential antiviral agents for SARS-CoV-2 using molecular hybridization approach. Molecules 2022, 27, 5923.
- Ghanim, A.M.; Girgis, A.S.; Kariuki, B.M.; Samir, N.; Said, M.F.; Abdelnaser, A.; Nasr, S.; Bekheit, M.S.; Abdelhameed, M.F.; Almalki, A.J.; et al. Design and synthesis of ibuprofen-quinoline conjugates as potential anti-inflammatory and analgesic drug candidates. Bioorg. Chem. 2022, 119, 105557.
- Panda, S.S.; Girgis, A.S.; Mishra, B.B.; Elagawany, M.; Devarapalli, V.; Littlefield, W.F.; Samir, A.; Fayad, W.; Fawzy, N.G.; Srour, A.M.; et al. Synthesis, computational studies, antimycobacterial and antibacterial properties of pyrazinoic acid-isoniazid hybrid conjugates. RSC Adv. 2019, 9, 20450–20462.
- Hein, C.D.; Liu, X.; Wang, D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 2008, 25, 2216–2230.
- Kaur, J.; Saxena, M.; Rishi, N. An overview of recent advances in biomedical applications of click chemistry. Bioconjugate Chem. 2021, 32, 1455–1471.
- Kondengadan, S.M.; Bansal, S.; Yang, C.; Liu, D.; Fultz, Z.; Wang, B. Click chemistry and drug delivery: A bird’s-eye view. Acta Pharm. Sin. B 2023, 13, 1990–2016.
- Devaraj, N.K.; Finn, M.G. Introduction: Click chemistry. Chem. Rev. 2021, 121, 6697–6698.
- Best, M.D. Click chemistry and bioorthogonal reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009, 48, 6571–6584.
- Bahia, S.B.B.B.; Reis, W.J.; Jardim, G.A.M.; Souto, F.T.; De Simone, C.A.; Gatto, C.C.; Menna-Barreto, R.F.; de Castro, S.L.; Cavalcanti, B.C.; Pessoa, C.; et al. Molecular hybridization as a powerful tool towards multitarget quinoidal systems: Synthesis, trypanocidal and antitumor activities of naphthoquinone-based 5-iodo-1,4-disubstituted-, 1,4- and 1,5-disubstituted-1,2,3-triazoles. Med. Chem. Commun. 2016, 7, 1555–1563.
- Thomopoulou, P.; Sachs, J.; Teusch, N.; Mariappan, A.; Gopalakrishnan, J.; Schmalz, H.G. New colchicine-derived triazoles and their influence on cytotoxicity and microtubule morphology. ACS Med. Chem. Lett. 2016, 7, 188–191.
- Li, H.-N.; Wang, H.; Wang, Z.-P.; Yan, H.-N.; Zhang, M.; Liu, Y.; Cheng, M.-S. Synthesis, antitumor activity evaluation and mechanistic study of novel hederacolchiside a1 derivatives bearing an aryl triazole moiety. Bioorg. Med. Chem. 2018, 26, 4025–4033.
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599.
- Sinha, S.; Kumaran, A.P.; Mishra, D.; Paira, P. Synthesis and cytotoxicity study of novel 3-(triazolyl)coumarins based fluorescent scaffolds. Bioorg. Med. Chem. Lett. 2016, 26, 5557–5561.
- Mbua, N.E.; Guo, J.; Wolfert, M.A.; Steet, R.; Boons, G.J. Strain-promoted alkyne-azide cycloadditions (SPAAC) reveal new features of glycoconjugate biosynthesis. ChemBioChem 2011, 12, 1912–1921.
- Kumar, G.S.; Qing, L. Light-Triggered Click Chemistry. Chem. Rev. 2021, 121, 6991–7031.
- Pérez-Balderas, F.; Ortega-Muñoz, M.; Morales-Sanfrutos, J.; Hernández-Mateo, F.; Calvo-Flores, F.G.; Calvo-Asín, J.A.; Isac-García, J.; Santoyo-González, F. Multivalent Neoglycoconjugates by Regiospecific Cycloaddition of Alkynes and Azides Using Organic-Soluble Copper Catalysts. Org. Lett. 2003, 5, 1951–1954.
- Arslan, M.; Acik, G.; Tasdelen, M.A. The emerging applications of click chemistry reactions in the modification of industrial polymers. Polym. Chem. 2019, 10, 3806–3821.
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028.
- Mehta, G.; Singh, V. Hybrid systems through natural product leads: An approach towards new molecular entities. Chem. Soc. Rev. 2002, 31, 324–334.
- Tomohara, K.; Ohashi, N.; Uchida, T.; Nose, T. Synthesis of natural product hybrids by the Ugi reaction in complex media containing plant extracts. Sci. Rep. 2022, 12, 15568.
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 2010, 110, 4489–4497.
- O’Connor, S.E.; Maresh, J.J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547.
- Faisal, S.; Badshah, S.L.; Kubra, B.; Emwas, A.; Jaremko, M. Alkaloids as potential antivirals. A comprehensive review. Nat. Prod. Bioprospect. 2023, 13, 4.
- Li, J.; Li, J.-X.; Jiang, H.; Li, M.; Chen, L.; Wang, Y.-Y.; Wang, L.; Zhang, N.; Guo, H.-Z.; Ma, K.-L. Phytochemistry and biological activities of corynanthe alkaloids. Phytochemistry 2023, 213, 113786.
- Wang, X.; Zhuang, Y.; Wang, Y.; Jiang, M.; Yao, L. The recent developments of camptothecin and its derivatives as potential anti-tumor agents. Eur. J. Med. Chem. 2023, 260, 115710.
- Islam, F.; Dehbia, Z.; Zehravi, M.; Das, R.; Sivakumar, M.; Krishnan, K.; Billah, A.A.M.; Bose, B.; Ghosh, A.; Paul, S.; et al. Indole alkaloids from marine resources: Understandings from therapeutic point of view to treat cancers. Chem. Biol. Interact. 2023, 383, 110682.
- Wang, L.; Jiang, Q.; Chen, S.; Wang, S.; Lu, J.; Gao, X.; Zhang, D.; Jin, X. Natural epidithiodiketopiperazine alkaloids as potential anticancer agents: Recent mechanisms of action, structural modification, and synthetic strategies. Bioorg. Chem. 2023, 137, 106642.
- Skiera, I.; Antoszczak, M.; Trynda, J.; Wietrzyk, J.; Boratynśki, P.; Kacprzak, K.; Huczynśki, A. Antiproliferative activity of polyether antibiotic—Cinchona alkaloid conjugates obtained via click chemistry. Chem. Biol. Drug Des. 2015, 86, 911–917.
- Wang, L.; Yuan, W.; Zhang, J.; Tong, L.; Luo, Y.; Chen, Y.; Lu, W.; Huang, Q. Synthesis of 7-triazole-substituted camptothecin via click chemistry and evaluation of in vitro antitumor activity. Chin. J. Chem. 2014, 32, 157–162.
- Nicolaus, N.; Zapke, J.; Riesterer, P.; Neudörfl, J.-M.; Prokop, A.; Oschkinat, H.; Schmalz, H.-G. azides derived from colchicine and their use in library synthesis: A practical entry to new bioactive derivatives of an old natural drug. Chem. Med. Chem. 2010, 5, 661–665.
- Kuznetsova, N.R.; Svirshchevskaya, E.V.; Sitnikov, N.S.; Abodo, L.; Sutorius, H.; Zapke, J.; Velder, J.; Thomopoulou, P.; Oschkinat, H.; Prokop, A.; et al. Lipophilic prodrugs of a triazole-containing colchicine analogue in liposomes: Biological effects on human tumor cells. Russ. J. Bioorg. Chem. 2013, 39, 543–552.
- Malysheva, Y.B.; Combes, S.; Allegro, D.; Peyrot, V.; Knochel, P.; Gavryushin, A.E.; Fedorov, A.Y. Synthesis and biological evaluation of novel anticancer bivalent colchicine-tubulizine hybrids. Bioorg. Med. Chem. 2012, 20, 4271–4278.
- Shi, A.; Huang, L.; Lu, C.; He, F.; Li, X. Synthesis, biological evaluation and molecular modeling of novel triazole-containing berberine derivatives as acetylcholinesterase and β-amyloid aggregation inhibitors. Bioorg. Med. Chem. 2011, 19, 2298–2305.
- Jin, X.; Yan, T.-H.; Yan, L.; Li, Q.; Wang, R.-L.; Hu, Z.-L.; Jiang, Y.-Y.; Sun, Q.-Y.; Cao, Y.-B. Design, synthesis, and anticancer activity of novel berberine derivatives prepared via cuaac ′′click′′ chemistry as potential anticancer agents. Drug. Des. Dev. Ther. 2014, 8, 1047–1059.
- Pingaew, R.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Design, synthesis and molecular docking studies of novel n-benzenesulfonyl-1,2,3,4-tetrahydroisoquinoline-based triazoles with potential anticancer activity. Eur. J. Med. Chem. 2014, 81, 192–203.
- Bunders, C.; Cavanagh, J.; Melander, C. flustramine inspired synthesis and biological evaluation of pyrroloindoline triazole amides as novel inhibitors of bacterial biofilms. Org. Biomol. Chem. 2011, 9, 5476–5481.
- Jiaranaikulwanitch, J.; Boonyarat, C.; Fokin, V.V.; Vajragupta, O. Triazolyl tryptoline derivatives as β-secretase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 6572–6576.
- Sangshetti, J.N.; Nagawade, R.R.; Shinde, D.B. synthesis of novel 3-(1-(1-substituted piperidin-4-yl)-1h-1,2,3-triazol-4-yl)-1,2,4- oxadiazol-5(4h)-one as antifungal agents. Bioorg. Med. Chem. Lett. 2009, 19, 3564–3567.
- Darandale, S.N.; Mulla, N.A.; Pansare, D.N.; Sangshetti, J.N.; Shinde, D.B. A novel amalgamation of 1,2,3-triazoles, piperidines and thieno pyridine rings and evaluation of their antifungal activity. Eur. J. Med. Chem. 2013, 65, 527–532.
- Corrales, R.C.N.R.; de Souza, N.B.; Pinheiro, L.S.; Abramo, C.; Coimbra, E.S.; Da Silva, A.D. Thiopurine derivatives containing triazole and steroid: Synthesis, antimalarial and antileishmanial activities. Biomed. Pharmacother. 2011, 65, 198–203.
- Nair, N.; Kudo, W.; Smith, M.A.; Abrol, R.; Goddard, W.A., III; Reddy, V.P. Novel purine-based fluoroaryl-1,2,3-triazoles as neuro- protecting agents: Synthesis, neuronal cell culture investigations, and cdk5 docking studies. Bioorg. Med. Chem. Lett. 2011, 21, 3957–3961.
- Wu, M.; Han, G.; Meng, C.; Wang, Z.; Liu, Y.; Wang, Q. Design, synthesis, and anti-tobacco mosaic virus (tmv) activity of glycoconjugates of phenanthroindolizidines alkaloids. Mol. Divers. 2014, 18, 25–37.
- Jin, C.; Navarro, H.A.; Carroll, F.I. Synthesis and receptor binding properties of 2β-alkynyl and 2β-(1,2,3-triazol)substituted 3β- (substituted phenyl)tropane derivatives. Bioorg. Med. Chem. 2008, 16, 5529–5535.
- Thuy, T.T.T.; Cuong, N.M.; Toan, T.Q.; Thang, N.N.; Tai, B.H.; Nhiem, N.X.; Hong, H.-J.; Kim, S.; Legoupy, S.; Koh, Y.S.; et al. Synthesis of novel derivatives of murrayafoline a and their inhibitory effect on lps-stimulated production of pro-inflammatory cytokines in bone marrow-derived dendritic cells. Arch. Pharma. Res. 2013, 36, 832–839.
- Zhao, L.; Mao, L.; Hong, G.; Yang, X.; Liu, T. Design, synthesis and anticancer activity of matrine-1h-1,2,3-triazole-chalcone conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 2540–2544.
- Li, S.; Li, X.-Y.; Zhang, T.-J.; Kamara, M.O.; Liang, J.-W.; Zhu, J.; Meng, F.-H. Design, synthesis and biological evaluation of homoerythrina alkaloid derivatives bearing a triazole moiety as parp-1 inhibitors and as potential antitumor drugs. Bioorg. Chem. 2020, 94, 103385.
- Li, S.; Li, X.-Y.; Zhang, T.J.; Zhu, J.; Xue, W.-H.; Qian, X.-H.; Meng, F.-H. Design, synthesis and biological evaluation of erythrina derivatives bearing a 1,2,3-triazole moiety as parp-1 inhibitors. Bioorg. Chem. 2020, 96, 103575.
- Artyushin, O.I.; Moiseeva, A.A.; Zarubaev, V.V.; Slita, A.V.; Galochkina, A.V.; Muryleva, A.A. Synthesis of camphecene and cytisine conjugates using click chemistry methodology and study of their antiviral activity. Chem. Biodivers. 2019, 16, e1900340.
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Tirumalasetty, M.C.B.; Wudayagiri, R.; Donthabakthuni, S. Design, synthesis, anticancer activity and docking studies of theophylline containing 1,2,3-triazoles with variant amide derivatives. Med. Chem. Commun. 2017, 8, 176–183.
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Chandra Babu Tirumalasetty, M.; Wudayagiri, R.; Donthabakthuni, S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396.
- Boratyński, P.J.; Gałęzowska, J.; Turkowiak, K.; Anisiewicz, A.; Kowalczyk, R.; Wietrzyk, J. Triazole biheterocycles fromcinchonaalkaloids: Coordination and antiproliferative properties. ChemistrySelect 2018, 3, 9368–9373.
- Podolski-Renić, A.; Bősze, S.; Dinić, J.; Kocsis, L.; Hudecz, F.; Csámpai, A.; Pešić, M. Ferrocene-cinchona hybrids with triazolyl-chalcone linkers act as pro-oxidants and sensitize human cancer cell lines to paclitaxel. Metallomics 2017, 9, 1132–1141.
- Sahu, A.; Agrawal, R.K.; Pandey, R. synthesis and systemic toxicity assessment of quinine-triazole scaffold with antiprotozoal potency. Bioorg. Chem. 2019, 88, 102939.
- Faidallah, H.M.; Panda, S.S.; Serrano, J.C.; Girgis, A.S.; Khan, K.A.; Alamry, K.A.; Therathanakorn, T.; Meyers, M.J.; Sverdrup, F.M.; Eickhoff, C.S.; et al. Synthesis, antimalarial properties and 2d-qsar studies of novel triazole-quinine conjugates. Bioorg. Med. Chem. 2016, 24, 3527–3539.
- Jin, X.; Yan, L.; Li, H.-j.; Wang, R.-L.; Hu, Z.-L.; Jiang, Y.-Y.; Cao, Y.-B.; Yan, T.-H.; Sun, Q.-Y. Novel triazolyl berberine derivatives prepared via cuaac click chemistry: Synthesis, anticancer activity and structure-activity relationships. Anticancer Agents Med. Chem. 2014, 15, 89–98.
- Batra, N.; Rajendran, V.; Agarwal, D.; Wadi, I.; Ghosh, P.C.; Gupta, R.D.; Nath, M. Synthesis and antimalarial evaluation of -triazole-tethered sulfonamide-berberine hybrids. ChemistrySelect 2018, 3, 9790–9793.
- Xu, X.; Wu, Y.; Liu, W.; Sheng, C.; Yao, J.; Dong, G.; Fang, K.; Li, J.; Yu, Z.; Min, X.; et al. Discovery of 7-methyl-10-hydroxyhomocamptothecins with 1,2,3-triazole moiety as potent topoisomerase I inhibitors. Chem. Biol. Drug Des. 2016, 88, 398–403.
- Konan, K.V.; Le, T.C.; Mateescu, M.A. Enhanced solubility of alkaloids by complexation with polycarboxylic materials for controlled release formulations: Case of peschiera fuchsiaefolia. AAPS PharmSciTech 2019, 20, 108.
- Alshamrani, M.; Khan, M.K.; Khan, B.A.; Salawi, A.; Almoshari, Y. Technologies for solubility, dissolution and permeation enhancement of natural compounds. Pharmaceuticals 2022, 15, 653.
- Venditti, A. Artifacts in natural products studies. An old and underestimated re-emerging problem. Nat. Prod. Res. 2018, 32, i–ii.
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204.
- Flores-Holguin, N.; Frau, J.; Glossman-Mitnik, D. computational pharmacokinetics report, admet study and conceptual dft-based estimation of the chemical reactivity properties of marine cyclopeptides. ChemistryOpen 2021, 10, 1142–1149.
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-score—A comprehensive scoring function for evaluation of chemical drug-likeness. MedChemComm 2019, 10, 148–157.
- Duran-Iturbide, N.A.; Diaz-Eufracio, B.I.; Medina-Franco, J.L. In silico adme/tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084.
- Zhang, X.; Liu, T.; Fan, X.; Ai, N. In silico modeling on ADME properties of natural products: Classification models for blood-brain barrier permeability, its application to traditional Chinese medicine and in vitro experimental validation. J. Mol. Graph. Model. 2017, 75, 347–354.
- Lin, J.; Sahakian, D.C.; de Morais, S.M.F.; Xu, J.J.; Polzer, R.J.; Winter, S.M. The role of absorption, distribution, metabolism, excretion and toxicity in drug discovery. Curr. Top. Med. Chem. 2003, 3, 1125–1154.
- Tibbitts, J.; Canter, D.; Graff, R.; Smith, A.; Khawli, L.A. Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. MAbs 2016, 8, 229–245.
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623.
- Mullard, A. Re-assessing the rule of 5, two decades on. Nat. Rev. Drug Discov. 2018, 17, 777.
- Gomis-Tena, J.; Brown, B.M.; Cano, J.; Trenor, B.; Yang, P.C.; Saiz, J.; Clancy, C.E.; Romero, L. When does the IC50 accurately assess the blocking potency of a drug? J. Chem. Inf. Model. 2020, 60, 1779–1790.
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972.
This entry is offline, you can click here to edit this entry!