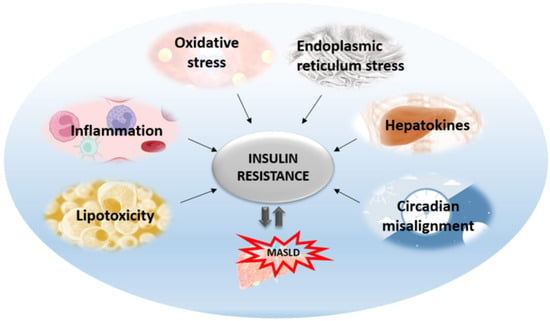
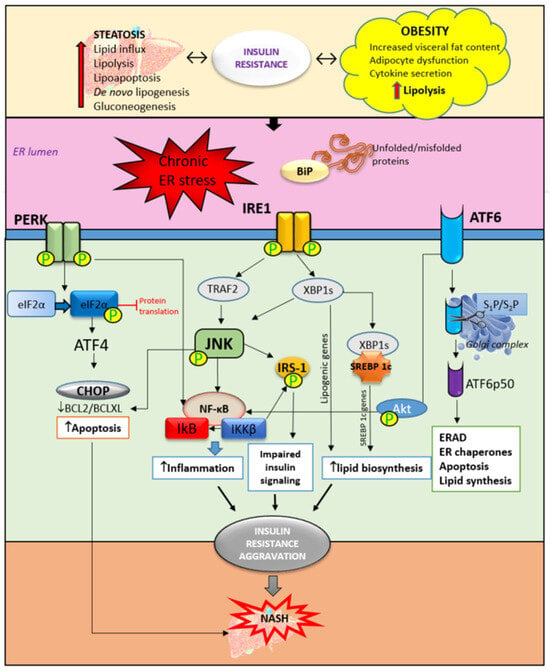
The central mechanism involved in the pathogenesis of MAFLD is insulin resistance with hyperinsulinemia, which stimulates triglyceride synthesis and accumulation in the liver. On the other side, triglyceride and free fatty acid accumulation in hepatocytes promotes insulin resistance via oxidative stress, endoplasmic reticulum stress, lipotoxicity, and the increased secretion of hepatokines. Cytokines and adipokines cause insulin resistance, thus promoting lipolysis in adipose tissue and ectopic fat deposition in the muscles and liver. Free fatty acids along with cytokines and adipokines contribute to insulin resistance in the liver via the activation of numerous signaling pathways. The secretion of hepatokines, hormone-like proteins, primarily by hepatocytes is disturbed and impairs signaling pathways, causing metabolic dysregulation in the liver. ER stress and unfolded protein response play significant roles in insulin resistance aggravation through the activation of apoptosis, inflammatory response, and insulin signaling impairment mediated via IRE1/PERK/ATF6 signaling pathways and the upregulation of SREBP 1c. Circadian rhythm derangement and biological clock desynchronization are related to metabolic disorders, insulin resistance, and NAFLD, suggesting clock genes as a potential target for new therapeutic strategies.
- insulin resistance
- NAFLD
- hepatokines
- ER stress
- circadian clock
- low-grade inflammation
- adipose tissue
- lipotoxicity
1. Introduction
2. Hepatic ER Stress and Unfolded Protein Response in Insulin Resistance
3. Insulin Resistance—From Adipose Tissue to the Liver
4. Lipid Accumulation in the Liver and Insulin Resistance
5. Hepatic Inflammation and Insulin Resistance
6. Liver Oxidative Stress and Insulin Resistance
7. The Role of Hepatokines in Insulin Resistance
This entry is adapted from the peer-reviewed paper 10.3390/cimb45110570
References
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081.
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619–639.
- Liu, C.; Zhou, B.; Meng, M.; Zhao, W.; Wang, D.; Yuan, Y.; Zheng, Y.; Qiu, J.; Li, Y.; Li, G.; et al. FOXA3 induction under endoplasmic reticulum stress contributes to non-alcoholic fatty liver disease. J. Hepatol. 2021, 75, 150–162.
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818.
- Lachkar, F.; Papaioannou, A.; Ferré, P.; Foufelle, F. Stress du réticulum endoplasmique et stéatopathies métaboliques . Biol. Aujourdhui 2020, 214, 15–23.
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.H.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J. ER stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2022, 43, 97–109.
- Petito-da-Silva, T.I.; Souza-Mello, V.; Barbosa-da-Silva, S. Empaglifozin mitigates NAFLD in high-fat-fed mice by alleviating insulin resistance, lipogenesis and ER stress. Mol. Cell Endocrinol. 2019, 498, 110539.
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695.
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843.
- Wu, X.; Xu, N.; Li, M.; Huang, Q.; Wu, J.; Gan, Y.; Chen, L.; Luo, H.; Li, Y.; Huang, X.; et al. Protective Effect of Patchouli Alcohol Against High-Fat Diet Induced Hepatic Steatosis by Alleviating Endoplasmic Reticulum Stress and Regulating VLDL Metabolism in Rats. Front. Pharmacol. 2019, 10, 1134.
- Xiao, T.; Liang, X.; Liu, H.; Zhang, F.; Meng, W.; Hu, F. Mitochondrial stress protein HSP60 regulates ER stress-induced hepatic lipogenesis. J. Mol. Endocrinol. 2020, 64, 67–75.
- Tian, T.; Liao, X.C.; Zhang, M.; Wu, X.M.; Guo, Y.T.; Tan, S.Y. Effects of celastrol on autophagy and endoplasmic reticulum stress-mediated apoptosis in a mouse model of nonalcoholic fatty liver disease. Chin. J. Hepatol. 2022, 30, 656–662.
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947.
- Wang, J.M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal 2018, 11, eaao4617.
- Mansour, S.Z.; Moustafa, E.M.; Moawed, F.S.M. Modulation of endoplasmic reticulum stress via sulforaphane-mediated AMPK upregulation against nonalcoholic fatty liver disease in rats. Cell Stress Chaperones 2022, 27, 499–511.
- Riaz, T.A.; Junjappa, R.P.; Handigund, M.; Ferdous, J.; Kim, H.-R.; Chae, H.-J. Role of Endoplasmic Reticulum Stress Sensor IRE1α in Cellular Physiology, Calcium, ROS Signaling, and Metaflammation. Cells 2020, 9, 1160.
- Choi, S.W.; Cho, W.; Oh, H.; Abd El-Aty, A.M.; Hong, S.A.; Hong, M.; Jeong, J.H.; Jung, T.W. Madecassoside ameliorates hepatic steatosis in high-fat diet-fed mice through AMPK/autophagy-mediated suppression of ER stress. Biochem. Pharmacol. 2023, 217, 115815.
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell Physiol. 2018, 233, 3867–3874.
- Moragrega, A.B.; Gruevska, A.; Fuster-Martínez, I.; Benedicto, A.M.; Tosca, J.; Montón, C.; Victor, V.M.; Esplugues, J.V.; Blas-García, A.; Apostolova, N. Anti-inflammatory and immunomodulating effects of rilpivirine: Relevance for the therapeutics of chronic liver disease. Biomed. Pharmacother. 2023, 167, 115537.
- Brown, M.; Dainty, S.; Strudwick, N.; Mihai, A.D.; Watson, J.N.; Dendooven, R.; Paton, A.W.; Paton, J.C.; Schröder, M. Endoplasmic reticulum stress causes insulin resistance by inhibiting delivery of newly synthesized insulin receptors to the cell surface. Mol. Biol. Cell 2020, 31, 2597–2629.
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827.
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315.
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000.
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877.
- da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607.
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806.
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135.
- Li, C.; Spallanzani, R.G.; Mathis, D. Visceral adipose tissue Tregs and the cells that nurture them. Immunol. Rev. 2020, 295, 114–125.
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2022, 45, 31–44.
- Park, S.H.; Liu, Z.; Sui, Y.; Helsley, R.N.; Zhu, B.; Powell, D.K.; Kern, P.A.; Zhou, C. IKKβ Is Essential for Adipocyte Survival and Adaptive Adipose Remodeling in Obesity. Diabetes 2016, 65, 1616–1629.
- Boutari, C.; Mantzoros, C.S. Adiponectin and leptin in the diagnosis and therapy of NAFLD. Metabolism 2020, 103, 154028.
- Shabalala, S.C.; Dludla, P.V.; Mabasa, L.; Kappo, A.P.; Basson, A.K.; Pheiffer, C.; Johnson, R. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed. Pharmacother. 2020, 131, 110785.
- Gastaldelli, A.; Gaggini, M.; DeFronzo, R.A. Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes 2017, 66, 815–822.
- Petrescu, M.; Vlaicu, S.I.; Ciumărnean, L.; Milaciu, M.V.; Mărginean, C.; Florea, M.; Vesa, Ș.C.; Popa, M. Chronic Inflammation-A Link between Nonalcoholic Fatty Liver Disease (NAFLD) and Dysfunctional Adipose Tissue. Medicina 2022, 58, 641.
- Munhoz, A.C.; Serna, J.D.C.; Vilas-Boas, E.A.; Caldeira da Silva, C.C.; Santos, T.G.; Mosele, F.C.; Felisbino, S.L.; Martins, V.R.; Kowaltowski, A.J. Adiponectin reverses β-Cell damage and impaired insulin secretion induced by obesity. Aging Cell 2023, 22, e13827.
- Bungau, S.; Behl, T.; Tit, D.; Banica, F.; Bratu, O.; Diaonu, C.; Nistor-Cseppento, C.; Bustea, C.; Corb Aron, R.A.; Vesa, C.M. Interactions between leptin and insulin resistance in patients with prediabetes, with and without NAFLD. Exp. Med. 2020, 20, 197.
- MacHado, M.V.; Coutinho, J.; Carepa, F.; Costa, A.; Proença, H.; Cortez-Pinto, H. How adiponectin, leptin, and ghrelin orchestrate together and correlate with the severity of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1166–1172.
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; Del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762.
- Saponaro, C.; Sabatini, S.; Gaggini, M.; Carli, F.; Rosso, C.; Positano, V.; Armandi, A.; Caviglia, G.P.; Faletti, R.; Bugianesi, E.; et al. Adipose tissue dysfunction and visceral fat are associated with hepatic insulin resistance and severity of NASH even in lean individuals. Liver Int. 2022, 42, 2418–2427.
- Guzzardi, M.A.; Hodson, L.; Guiducci, L.; La Rosa, F.; Salvadori, P.A.; Burchielli, S.; Iozzo, P. The role of glucose, insulin and NEFA in regulating tissue triglyceride accumulation: Substrate cooperation in adipose tissue versus substrate competition in skeletal muscle. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 956–963.
- Abel, E.D.; Peroni, O.D.; Kim, J.; Kim, Y.-B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733.
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 2005, 54, 3458–3465.
- Pal, S.C.; Méndez-Sánchez, N. Insulin resistance and adipose tissue interactions as the cornerstone of metabolic (dysfunction)-associated fatty liver disease pathogenesis. World J. Gastroenterol. 2023, 29, 3999–4008.
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048.
- Ferramosca, A.; Zara, V. Modulation of hepatic steatosis by dietary fatty acids. World J. Gastroenterol. 2014, 20, 1746.
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374.
- George, J.; Liddle, C. Nonalcoholic Fatty Liver Disease: Pathogenesis and Potential for Nuclear Receptors as Therapeutic Targets. Mol. Pharm. 2008, 5, 49–59.
- Gao, B.; Tsukamoto, H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016, 150, 1704–1709.
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203.
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of lipotoxicity in NAFLD and clinical implications. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 131–140.
- Ponziani, F.R.; Pecere, S.; Gasbarrini, A.; Ojetti, V. Physiology and pathophysiology of liver lipid metabolism. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1055–1067.
- Jaganjac, M.; Milkovic, L.; Zarkovic, N.; Zarkovic, K. Oxidative stress and regeneration. Free Radic. Biol. Med. 2022, 181, 154–165.
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology 2006, 130, 1245–1258.
- Hubbard, B.; Doege, H.; Punreddy, S.; Wu, H.; Huang, X.; Kaushik, V.K.; Mozell, R.L.; Byrnes, J.J.; Stricker-Krongrad, A.; Chou, C.J.; et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology 2006, 130, 1259–1269.
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192.
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.B.; Girard, J.; Postic, C. Liver-Specific Inhibition of ChREBP Improves Hepatic Steatosis and Insulin Resistance in ob/ob Mice. Diabetes 2006, 55, 2159–2170.
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189.
- Yao, M.; Zhou, P.; Qin, Y.Y.; Wang, L.; Yao, D.F. Mitochondrial carnitine palmitoyltransferase-II dysfunction: A possible novel mechanism for nonalcoholic fatty liver disease in hepatocarcinogenesis. World J. Gastroenterol. 2023, 29, 1765–1778.
- Orellana-Gavaldà, J.M.; Herrero, L.; Malandrino, M.I.; Pañeda, A.; Sol Rodríguez-Peña, M.; Petry, H.; Asins, G.; Van Deventer, S.; Hegardt, F.G.; Serra, D. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology 2011, 53, 821–832.
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353.
- Stanković, M.N.; Mladenović, D.R.; Duričić, I.; Šobajić, S.S.; Timić, J.; Jorgačević, B.; Aleksić, V.; Vučević, D.B.; Ješić-Vukićević, R.; Radosavljević, T.S. Time-dependent changes and association between liver free fatty acids, serum lipid profile and histological features in mice model of nonalcoholic fatty liver disease. Arch. Med. Res. 2014, 45, 116–124.
- Veskovic, M.; Mladenovic, D.; Milenkovic, M.; Tosic, J.; Borozan, S.; Gopcevic, K.; Labudovic-Borovic, M.; Dragutinovic, V.; Vucevic, D.; Jorgacevic, B.; et al. Betaine modulates oxidative stress, inflammation, apoptosis, autophagy, and Akt/mTOR signaling in methionine-choline deficiency-induced fatty liver disease. Eur. J. Pharmacol. 2019, 848, 39–48.
- Vesković, M.; Labudović-Borović, M.; Mladenović, D.; Jadžić, J.; Jorgačević, B.; Vukićević, D.; Vučević, D.; Radosavljević, T. Effect of Betaine Supplementation on Liver Tissue and Ultrastructural Changes in Methionine-Choline-Deficient Diet-Induced NAFLD. Microsc. Microanal. 2020, 26, 997–1006.
- Liang, W.; Lindeman, J.H.; Menke, A.L.; Koonen, D.P.; Morrison, M.; Havekes, L.M.; van den Hoek, A.M.; Kleemann, R. Metabolically induced liver inflammation leads to NASH and differs from LPS- or IL-1beta-induced chronic inflammation. Lab. Investig. 2014, 94, 491–502.
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2022, 65, 1064.
- Mohammed, E.D.; Zhang, Z.; Tian, W.; Gangarapu, V.; Al-Gendy, A.A.; Chen, J.; Wei, J.; Sun, B. Modulation of IR as a therapeutic target to prevent NASH using NRF from Diceratella elliptica (DC.) jonsell. Strong Nrf2 and leptin inducer as well as NF-kB inhibitor. Phytomedicine 2021, 80, 153388.
- Ren, F.; Zhou, L.; Zhang, X.; Wen, T.; Shi, H.; Xie, B.; Li, Z.; Chen, D.; Wang, Z.; Duan, Z. Endoplasmic reticulum stress-activated glycogen synthase kinase 3β aggravates liver inflammation and hepatotoxicity in mice with acute liver failure. Inflammation 2015, 38, 1151–1165.
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604.
- Gasmi, A.; Noor, S.; Menzel, A.; Doşa, A.; Pivina, L.; Bjørklund, G. Obesity and Insulin Resistance: Associations with Chronic Inflammation, Genetic and Epigenetic Factors. Curr. Med. Chem. 2021, 28, 800–826.
- Jung, T.W.; Park, H.S.; Choi, G.H.; Kim, D.; Lee, T. β-aminoisobutyric acid attenuates LPS-induced inflammation and insulin resistance in adipocytes through AMPK-mediated pathway. J. Biomed. Sci. 2018, 25, 27.
- Zhang, L.; Bansal, M.B. Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front. Immunol. 2020, 11, 1086.
- Gruben, N.; Shiri-Sverdlov, R.; Koonen, D.P.; Hofker, M.H. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochem. Biophys. Acta 2014, 1842, 2329–2343.
- Jorgačević, B.; Vučević, D.; Samardžić, J.; Mladenović, D.; Vesković, M.; Vukićević, D.; Ješić, R.; Radosavljević, T. The Effect of CB1 Antagonism on Hepatic Oxidative/Nitrosative Stress and Inflammation in Nonalcoholic Fatty Liver Disease. Curr. Med. Chem. 2021, 28, 169–180.
- Jorgačević, B.; Mladenović, D.; Ninković, M.; Vesković, M.; Dragutinović, V.; Vatazević, A.; Vučević, D.; Ješić Vukićević, R.; Radosavljević, T. Rimonabant Improves Oxidative/Nitrosative Stress in Mice with Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell Longev. 2015, 2015, 842108.
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128.
- Gurzov, E.N.; Tran, M.; Fernandez-Rojo, M.A.; Merry, T.L.; Zhang, X.; Xu, Y.; Fukushima, A.; Waters, M.J.; Watt, M.J.; Andrikopoulos, S.; et al. Hepatic oxidative stress promotes insulin-STAT-5 signaling and obesity by inactivating protein tyrosine phosphatase N2. Cell Metab. 2014, 20, 85–102.
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262.
- Ma, J.; Nakagawa, Y.; Kojima, I.; Shibata, H. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J. Biol. Chem. 2013, 298, 133–142.
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, 330–341.
- Tsai, H.; Wang, W.; Lin, C.; Pai, P.; Lai, T.; Tsai, C. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK dependent activation of NF-kB in cardiomyocytes exposed to high glucose. J. Cell Physiol. 2012, 227, 1347–1357.
- Al-Lahham, R.; Deford, J.; Papaconstantinou, J. Mitochondrial-generated ROS down regulates insulin signaling via activation of p38 MAPK stress response pathway. Mol. Cell Endocrinol. 2015, 419, 1–11.
- Polce, S.A.; Burke, C.; França, L.M.; Kramer, B.; de Andrade Paes, A.M.; Carrillo-Sepulveda, M.A. Ellagic Acid Alleviates Hepatic Oxidative Stress and Insulin Resistance in Diabetic Female Rats. Nutrients 2018, 10, 531.
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karinin, M. IKK-beta links inflammation to obesity induced insulin resistance. Nat. Med. 2005, 11, 191–198.
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 2021, 44, 101138.
- Jialal, I.; Pahwa, R. Fetuin-A is also an adipokine. Lipids Health Dis. 2019, 18, 73.
- Lanthier, N.; Lebrun, V.; Molendi-Coste, O.; van Rooijen, N.; Leclercq, I.A. Liver Fetuin-A at Initiation of Insulin Resistance. Metabolites 2022, 12, 1023.
- Yamasandhi, P.G.; Dharmalingam, M.; Balekuduru, A. Fetuin-A in newly detected type 2 diabetes mellitus as a marker of non-alcoholic fatty liver disease. Indian J. Gastroenterol. 2021, 40, 556–562.
- Sardana, O.; Goyal, R.; Bedi, O. Molecular and pathobiological involvement of fetuin-A in the pathogenesis of NAFLD. Inflammopharmacology 2021, 29, 1061–1074.
- Pagan, L.U.; Gatto, M.; Martinez, P.F.; Okoshi, K.; Okoshi, M.P. Biomarkers in Cardiovascular Disease: The Role of Fetuin-A. Arq. Bras. Cardiol. 2022, 118, 22–23.
- Chekol Abebe, E.; Tilahun Muche, Z.; Behaile, T.; Mariam, A.; Mengie Ayele, T.; Mekonnen Agidew, M.; Teshome Azezew, M.; Abebe Zewde, E.; Asmamaw Dejenie, T.; Asmamaw Mengstie, M. The structure, biosynthesis, and biological roles of fetuin-A: A review. Front. Cell Dev. Biol. 2022, 10, 945287.
- Kothari, V.; Babu, J.R.; Mathews, S.T. AMP activated kinase negatively regulates hepatic Fetuin-A via p38 MAPK-C/EBPβ/E3 Ubiquitin Ligase Signaling pathway. PLoS ONE 2022, 17, e0266472.
- Ren, G.; Kim, T.; Papizan, J.B.; Okerberg, C.K.; Kothari, V.M.; Zaid, H.; Bilan, P.J.; Araya-Ramirez, F.; Littlefield, L.A.; Bowers, R.L.; et al. Phosphorylation status of fetuin-A is critical for inhibition of insulin action and is correlated with obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E250–E260.
- Lee, K.Y.; Lee, W.; Jung, S.H.; Park, J.; Sim, H.; Choi, Y.J.; Park, Y.J.; Chung, Y.; Lee, B.H. Hepatic upregulation of fetuin-A mediates acetaminophen-induced liver injury through activation of TLR4 in mice. Biochem. Pharmacol. 2019, 166, 46–55.
- Mathews, S.T.; Singh, G.P.; Ranalletta, M.; Cintron, V.J.; Qiang, X.; Goustin, A.S.; Jen, K.L.; Charron, M.J.; Jahnen-Dechent, W.; Grunberger, G. Improved insulin sensitivity and resistance to weight gain in mice null for the Ahsg gene. Diabetes 2002, 51, 2450–2458.
- Zhou, W.; Yang, J.; Zhu, J.; Wang, Y.; Wu, Y.; Xu, L.; Yang, Y. Fetuin B aggravates liver X receptor-mediated hepatic steatosis through AMPK in HepG2 cells and mice. Am. J. Transl. Res. 2019, 11, 1498–1509.
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.Y.; Burke, M.; Goode, R.J.A.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015, 22, 1078–1089.
- Mokou, M.; Yang, S.; Zhan, B.; Geng, S.; Li, K.; Yang, M.; Yang, G.; Deng, W.; Liu, H.; Liu, D.; et al. Elevated Circulating Fetuin-B Levels Are Associated with Insulin Resistance and Reduced by GLP-1RA in Newly Diagnosed PCOS Women. Mediators Inflamm. 2020, 2020, 2483435.
- Wang, D.; Wu, M.; Zhang, X.; Li, L.; Lin, M.; Shi, X.; Zhao, Y.; Huang, C.; Li, X. Hepatokine Fetuin B expression is regulated by leptin-STAT3 signaling and associated with leptin in obesity. Sci. Rep. 2022, 12, 12869.
- Almarashda, O.; Abdi, S.; Yakout, S.; Khattak, M.N.K.; Al-Daghri, N.M. Hepatokines Fetuin-A and Fetuin-B status in obese Saudi patient with diabetes mellitus type 2. Am. J. Transl. Res. 2022, 14, 3292–3302.
- Peter, A.; Kovarova, M.; Staiger, H.; Machann, J.; Schick, F.; Königsrainer, A.; Königsrainer, I.; Schleicher, E.; Fritsche, A.; Häring, H.; et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E266–E273.
- Staiger, H.; Keuper, M.; Berti, L.; Hrabe de Angelis, M.; Häring, H. Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr. Rev. 2017, 38, 468–488.
- Kucukoglu, O.; Sowa, J.; Mazzolini, G.D.; Syn, W.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457.
- Urraza-Robledo, A.I.; Giralt, M.; González-Galarza, F.F.; Villarroya, F.; Miranda Pérez, A.A.; Ruiz Flores, P.; Gutiérrez Pérez, M.E.; Domingo, P.; López-Márquez, F.C. FGF21 serum levels are related to insulin resistance, metabolic changes and obesity in Mexican people living with HIV (PLWH). PLoS ONE 2021, 16, e0252144.
- Keinicke, H.; Sun, G.; Mentzel, C.M.J.; Fredholm, M.; John, L.M.; Andersen, B.; Raun, K.; Kjaergaard, M. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocr. Connect 2020, 9, 755–768.
- BonDurant, L.D.; Ameka, M.; Naber, M.C.; Markan, K.R.; Idiga, S.O.; Acevedo, M.R.; Walsh, S.A.; Ornitz, D.M.; Potthoff, M.J. FGF21 regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab. 2017, 25, 935–944.e4.
- Szczepańska, E.; Gietka-Czernel, M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm. Metab. Res. 2022, 54, 203–211.
- Pothoff, M.J.; Inagaki, T.; Satapati, S.; Ding, X.; He, T.; Goetz, R.; Mohammadi, M.; Finck, B.N.; Mangelsdorf, D.J.; Kliewer, S.A.; et al. FGF21 induces PGC-1a and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA 2009, 106, 10853–10858.
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936.
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807.
- Hariharan, S.; Dharmaraj, S. Selenium and selenoproteins: It’s role in regulation of inflammation. Inflammopharmacology 2020, 28, 667–695.
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495.
- Choi, H.Y.; Hwang, S.Y.; Lee, C.H.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Increased selenoprotein p levels in subjects with visceral obesity and nonalcoholic Fatty liver disease. Diabetes Metab. J. 2013, 37, 63–71.
- Yang, S.J.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: Implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metab. 2011, 96, 1325.
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393.
- Mita, Y.; Nakayama, K.; Inari, S.; Nishito, Y.; Yoshioka, Y.; Sakai, N.; Sotani, K.; Nagamura, T.; Kuzuhara, Y.; Inagaki, K.; et al. Selenoprotein P-neutralizing antibodies improve insulin secretion and glucose sensitivity in type 2 diabetes mouse models. Nat. Commun. 2017, 8, 1658.