Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
C–H bond functionalisation describes the transformation of a C-H bonds into a C-X bonds (X = C, N, O, B, halides, among many others). The term is typically associated with transition metals in processes like C-H activation. However, other methods such as nitrene/carbene insertion and hydrogen atom transfer (HAT) can also be used to functionalise C-H bonds with varying selectivity and ability to introduce different functional groups depending on the method used.
- hydrogen atom transfer
- functionalisation
- radicals
- photoredox
- electrochemistry
1. Introduction
The ability to use C–H bonds as de facto functional handles has streamlined the synthesis of complex organic molecules and changed how chemists approach retrosynthesis [1][2][3][4][5]. Using C–H bonds as functional handles instead of pre-functionalised substrates, the yields’ various benefits include lower step counts in multistep synthesis and an improved atom economy of reactions [3][6][7][8]. Furthermore, the diversity of transformations available to C–H bonds potentially enables access to a wide array of functionality to be introduced into a common core [7][9][10]. Broadly speaking, C–H functionalisation is achieved by generating reactive intermediates from C–H bonds to subsequently harness their reactivity. This can be achieved through organometallic C–H activation [6][11][12][13][14], carbene/nitrene C–H insertion [2][11][15][16][17], enzymatic C–H functionalisation [18], or hydrogen atom transfer (HAT) [11][19][20]. However, site-selective C–H functionalisation is challenging due to the minimal differences between C–H bonds in organic molecules [6][21][22][23]. HAT generates alkyl radicals from C–H bonds through the radical abstraction of hydrogen atoms [19]. Alkyl radicals are highly reactive intermediates that are relatively insensitive to steric crowding and do not form aggregates [24]. Alkyl radicals react chemoselectively with radical traps or couple with other radicals, even with substrates that contain N-heterocycles as well as polar and acidic functional groups [20][24][25][26][27][28]. Additionally, HAT processes can be fine-tuned towards specific C–H bonds through choice of HAT reagent, change of solvent, or addition of certain additives [29][30].
2. HAT Background and Mechanism
Hydrogen atom transfer is a one-step process that transfers a hydrogen atom (proton and electron) from one species to another (Scheme 1) [31][32]. However, in the context of synthesis, HAT is used for C–H functionalisation by harnessing the reactivity of the alkyl radical with various radical traps [19].
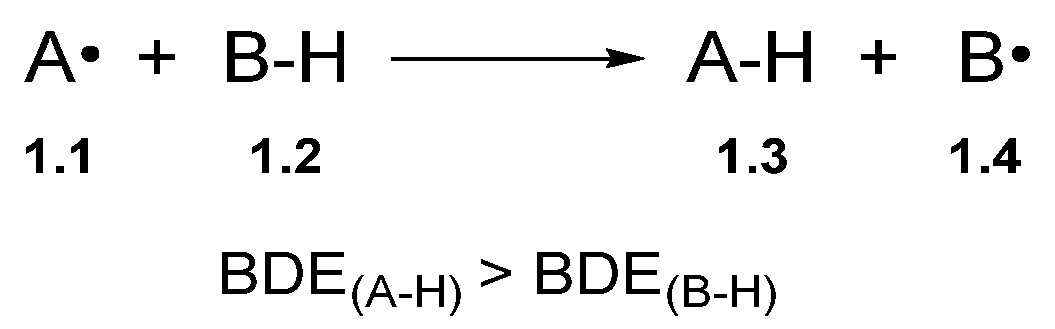
Scheme 1. Generic HAT process.
The bond dissociation energy (BDE) is the key driving force for HAT [33][34][35]. Accordingly, the BDE of A–H in Compound 1.3 (A–H) should be greater than the C–H bond being abstracted (B–H) to favour product formation [32]. Fortunately, BDE values are well-documented in the chemical literature [33][36][37]. BDE values can also be matched to ensure the desired hydrogen atom is abstracted. For example, thiyl radicals undergo HAT with relatively weak C–H bonds to form the corresponding alkyl radical and a thiol [alkyl thiols BDES–H ≈ 87 kcal/mol] [38]. Hamashima and co-workers developed an arylation of benzylamine 2.1 C(sp3)–H bonds, which proceeded through regio- and chemo-selective HAT of the benzylic C(sp3)–H using a thiyl radical derived from thiobenzoic acid 2.3 [N,N-dimethylbenzylamine 2.1 BDEC–H = 84.9 kcal/mol versus thiobenzoic acid 2.3 BDES–H = 87.4 kcal/mol] (Scheme 2) [39].

Scheme 2. Benzylamine C–H arylation using a thiyl radical formed from thiobenzoic acid.
Conversely, stronger HAT reagents can be used to abstract unactivated C(sp3)–H bonds of alkanes [33]. Knowles and co-workers reported the alkylation of cyclohexane 3.1 through HAT with amidyl radical 3.3• [amide 3.3 BDEN–H = 107 kcal/mol versus cyclohexane 3.1 BDEC–H = 99.5 kcal/mol] (Scheme 3) [40]. The high BDE value of N–H bonds in amides allows for HAT of unactivated C(sp3)–H bonds of alkanes. Hence, BDE values of the HAT reagent and the substrate should be matched carefully to ensure the HAT process is thermodynamically spontaneous and selective [33].
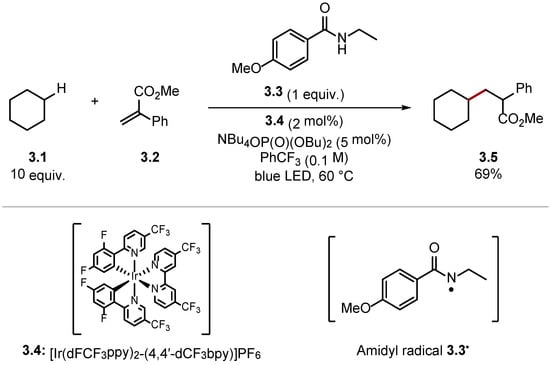
Scheme 3. C(sp3)–H alkylation using amide 3.3 as a HAT reagent.
Despite radicals being electronically neutral species, electronic factors in transition states of radical reactions greatly influence their rate and selectivity [41][42][43][44]. In this context, polar effects describe the effect the charge transfer has on the activation energy, and HAT is strongly influenced by such electronic factors. Usually, polarity-matched HAT describes the tendency of electrophilic HAT reagents to abstract electron-rich (“hydridic”) hydrogen atoms and nucleophilic HAT reagents to abstract electron-poor (“protic”) hydrogen atoms (Scheme 4) [41][45].
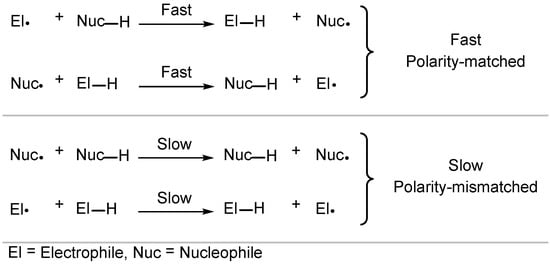
Scheme 4. Polarity effects in HAT processes.
Most HAT reagents are electrophilic and selectively abstract electron-rich hydrogen atoms [30]. As a result, HAT normally occurs adjacent to an electron-donating group (EDG) or another stabilizing functional group [19][46]. Bietti and co-workers have extensively studied the reaction rates of HAT [29][30][37][47][48][49][50][51][52][53][54][55][56][57][58][59][60]. Recently, Bietti studied the rates of HAT for saturated N-containing heterocycles and tetramethyl urea 5.1 using dicumyl peroxide 5.2 (Scheme 5) [47].
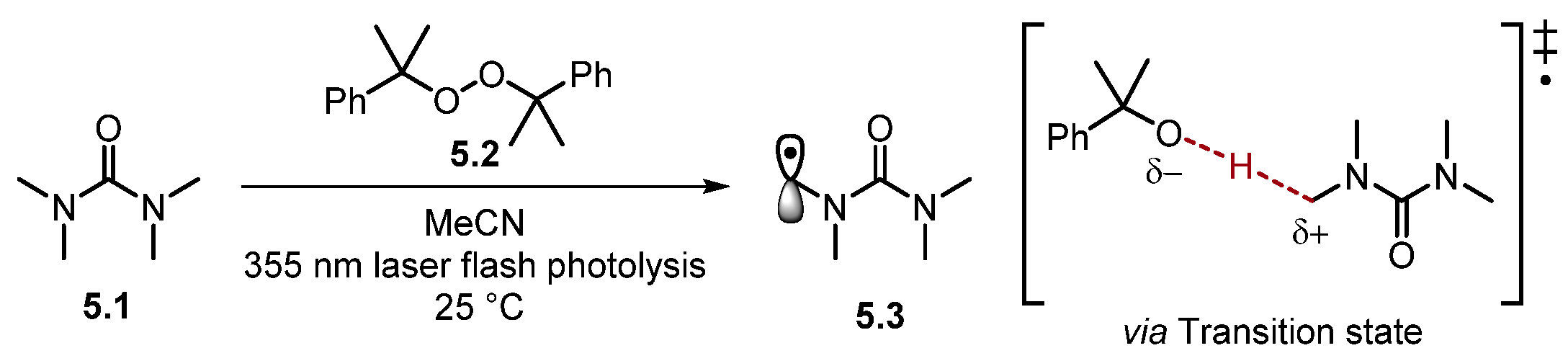
Scheme 5. Transition state (‡) of HAT with cumyloxyl radical.
The HAT transition state can be described as developing a partial positive charge at the C–atom, along with a partial negative charge on the abstracting radical (cumyloxyl radical in this case) [29][34][35]. Functional groups such as amides stabilise the partial positive charge on the incipient radical atom through an orbital overlap of the σ* of the α-C–H (developing SOMO) with a heteroatom lone pair or a π-system [47]. This transition state model has been probed through experimental observations, Hammett plot analysis, and computational studies [35][45][48][61]. HAT processes are dictated by an electron density at different C–H bonds meaning a change in solvent, an addition of H-bond donor/acceptors, or Brønsted/Lewis-acid/base additives can alter the rates of HAT [29][30]. For instance, strong H-bonding solvents [such as hexafluoroisopropanol (HFIP)] are used to supress undesired HAT adjacent to H-bond acceptors (e.g., heteroatoms) [50][62][63][64][65]. However, H-bonding solvents are also known to accelerate HAT at cyclohexane and 1,4-cyclohexadiene through H-bonding to oxyl-radicals [29][66][67][68].
The abstraction of protic hydrogen atoms is difficult as most HAT-capable radicals are inherently electrophilic heteroatom-centred radicals or radical cations [30]. However, the abstraction of protic hydrogens can be accomplished through a polarity reversal catalysis (PRC) [41]. PRC can generate a nucleophilic HAT reagent through an initial polarity-matched HAT step (Scheme 6) [69][70]. For example, electrophilic alkoxyl radical 6.2 reacts with amino boranes 6.3 to form a nucleophilic amine boryl radical 6.5, which abstracts protic hydrogen atoms selectively, such as acetonitrile 6.6 α-C(sp3)–H to generate electrophilic alkyl radical 6.7.
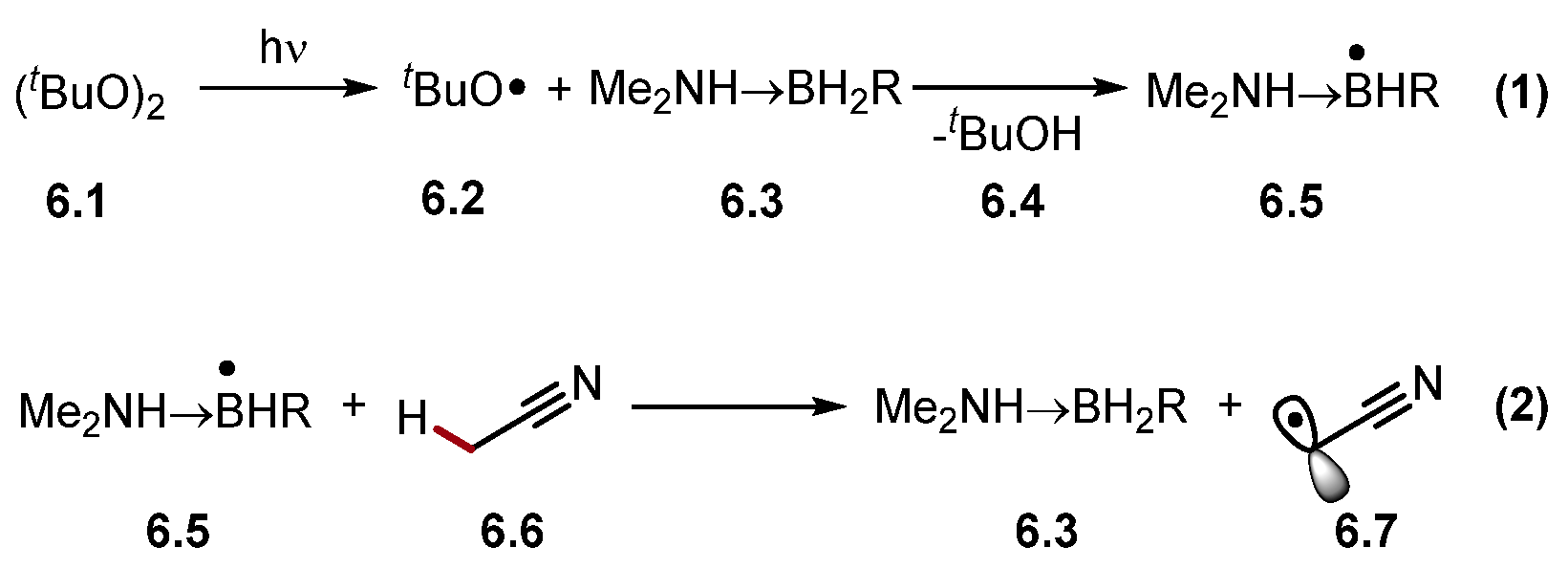
Scheme 6. HAT of protic hydrogen atom from MeCN using PRC.
3. Indirect HAT
As mentioned before, indirect HAT describes protocols where a radical H-atom abstractor is generated in situ [19][71]. Radicals capable of HAT are typically formed in the following ways (Scheme 7).

Scheme 7. Methods of generating a HAT-capable species in situ.
(1) Homolytic/Heterolytic cleavage of a weak bond. The weak O–O bond in peroxides 7.1 undergoes homolysis to form two oxygen-centred radicals 7.2 capable of HAT [tBuCH2O–OCH2tBu BDEO–O = 36.4 kcal/mol] [36][72]. The peroxide 7.1 can also undergo heterolysis with a reducing agent/acid to form one equivalent of O-centred radical 7.2 [72].
(2) Mesolytic cleavage of a radical ion. For instance, a redox active ester (RAE) 7.6 can be reduced to a radical anion [73][74][75][76]. The radical anion undergoes mesolytic cleavage forming CO2 7.7, phthalimide anion 7.9, and a methyl radical 7.8, which is a competent HAT reagent [77].
(3) Oxidation of a heteroatom or an anion using photoredox catalysis or (less commonly) electrochemistry [78]. For example, a thiolate 7.11 can be oxidised to a thiyl radical 7.12, which can abstract a hydrogen atom to form a thiol 7.13 and alkyl radical 7.5 [79]. Deprotonation of the thiol 7.13 allows the turnover of the HAT reagent to make the process catalytic.
(4) Radical propagation steps can continuously regenerate the HAT reagent (otherwise known as chain transfer). In the provided example, a fluorine atom transfer (FAT) between alkyl radical 7.5 and Selectfluor 7.14 affords the fluorinated product 7.16 and generates an equivalent of TEDA2+· 7.15· for further HAT [80]. Notably, chain transfer can also be a contributing pathway in reactions where Methods (1), (2), and (3) are the main pathways with widely varying degrees of chain contribution. In photoredox chemistry, the contribution of the chain transfer to the reaction mechanism can be investigated using quantum-yield measurements or “light/dark experiments” [81]. However, reactions that utilise chain propagation as the major pathway typically use a sub-stoichiometric amount of initiator to initiate the process [82][83][84][85][86].
Each method can form a radical species capable of HAT, therefore facilitating the generation of radicals from corresponding C–H bonds, as well as X–H bonds (where X = heteroatom). Harnessing the high reactivity of radicals allows for a multitude of transformations [28][33][87][88][89][90].
This entry is adapted from the peer-reviewed paper 10.3390/molecules28166127
References
- Lam, N.Y.S.; Wu, K.; Yu, J.-Q. Advancing the Logic of Chemical Synthesis: C–H Activation as Strategic and Tactical Disconnections for C–C Bond Construction. Angew. Chem. Int. Ed. 2021, 60, 15767–15790.
- Chu, J.C.K.; Rovis, T. Complementary Strategies for Directed C(sp3)–H Functionalisation: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed. 2018, 57, 62–101.
- Abrams, D.J.; Provencher, P.A.; Sorensen, E.J. Recent applications of C–H functionalisation in complex natural product synthesis. Chem. Soc. Rev. 2018, 47, 8925–8967.
- Yoshioka, S.; Nagatomo, M.; Inoue, M. Application of Two Direct C(sp3)–H Functionalisations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93.
- Falcone, N.A.; Bosse, A.T.; Park, H.; Yu, J.-Q.; Davies, H.M.L.; Sorensen, E.J. A C–H Functionalization Strategy Enables an Enantioselective Formal Synthesis of (–)-Aflatoxin B2. Org. Lett. 2021, 23, 9393–9397.
- Rogge, T.; Kaplaneris, N.; Chatani, N.; Kim, J.; Chang, S.; Punji, B.; Schafer, L.L.; Musaev, D.G.; Wencel-Delord, J.; Roberts, C.A.; et al. C–H activation. Nat. Rev. Methods Primers 2021, 1, 43.
- Capaldo, L.; Quadri, L.L.; Ravelli, D. Photocatalytic hydrogen atom transfer: The philosopher’s stone for late-stage functionalisation? Green Chem. 2020, 22, 3376–3396.
- Barham, J.P.; John, M.P.; Murphy, J.A. Contra-thermodynamic Hydrogen Atom Abstraction in the Selective C–H Functionalisation of Trialkylamine N-CH3 Groups. J. Am. Chem. Soc. 2016, 138, 15482–15487.
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalisation of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576.
- Candish, L.; Collins, K.D.; Cook, G.C.; Douglas, J.J.; Gómez-Suárez, A.; Jolit, A.; Keess, S. Photocatalysis in the Life Science Industry. Chem. Rev. 2022, 122, 2907–2980.
- Karimov, R.R.; Hartwig, J.F. Transition-Metal-Catalyzed Selective Functionalisation of C(sp3)–H Bonds in Natural Products. Angew. Chem. Int. Ed. 2018, 57, 4234–4241.
- Britton, L.; Docherty, J.; Sklyaruk, J.; Cooney, J.; Nichol, G.S.; Dominey, A.; Thomas, S.P. Iron-catalysed Alkene and Heteroarene H/D Exchange by Reversible Protonation of Iron-hydride Intermediates. Chem. Sci. 2022, 13, 10291–10298.
- Britton, L.; Skrodzki, M.; Nichol, G.S.; Dominey, A.P.; Pawluć, P.; Docherty, J.H.; Thomas, S.P. Manganese-Catalyzed C(sp2)–H Borylation of Furan and Thiophene Derivatives. ACS Catal. 2021, 11, 6857–6864.
- Britton, L.; Docherty, J.H.; Dominey, A.P.; Thomas, S.P. Iron-Catalysed C(sp2)–H Borylation Enabled by Carboxylate Activation. Molecules. 2020, 25, 905.
- He, Y.; Huang, Z.; Wu, K.; Ma, J.; Zhou, Y.-G.; Yu, Z. Recent advances in transition-metal-catalyzed carbene insertion to C–H bonds. Chem. Soc. Rev. 2022, 51, 2759–2852.
- Han, F.; Choi, P.H.; Ye, C.-X.; Grell, Y.; Xie, X.; Ivlev, S.I.; Chen, S.; Meggers, E. Cyclometalated Chiral-at-Ruthenium Catalyst for Enantioselective Ring-Closing C(sp3)–H Carbene Insertion to Access Chiral Flavanones. ACS Catal. 2022, 12, 10304–10312.
- Peil, S.; Gutiérrez González, A.; Leutzsch, M.; Fürstner, A. C–H Insertion via Ruthenium Catalyzed gem-Hydrogenation of 1,3-Enynes. J. Am. Chem. Soc. 2022, 144, 4158–4167.
- Romero, E.; Jones, B.S.; Hogg, B.N.; Rué Casamajo, A.; Hayes, M.A.; Flitsch, S.L.; Turner, N.J.; Schnepel, C. Enzymatic Late-Stage Modifications: Better Late Than Never. Angew. Chem. Int. Ed. 2021, 60, 16824–16855.
- Capaldo, L.; Ravelli, D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071.
- Golden, D.L.; Suh, S.-E.; Stahl, S.S. Radical C(sp3)–H functionalisation and cross-coupling reactions. Nat. Rev. Chem. 2022, 6, 405–427.
- Newhouse, T.; Baran, P.S. If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374.
- Feng, K.; Quevedo, R.E.; Kohrt, J.T.; Oderinde, M.S.; Reilly, U.; White, M.C. Late-stage oxidative C(sp3)–H methylation. Nature. 2020, 580, 621–627.
- Zhang, T.; Luan, Y.-X.; Lam, N.Y.S.; Li, J.-F.; Li, Y.; Ye, M.; Yu, J.-Q. A directive Ni catalyst overrides conventional site selectivity in pyridine C–H alkenylation. Nat. Chem. 2021, 13, 1207–1213.
- Yan, M.; Lo, J.C.; Edwards, J.T.; Baran, P.S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714.
- Crespi, S.; Fagnoni, M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833.
- Leifert, D.; Studer, A. The Persistent Radical Effect in Organic Synthesis. Angew. Chem. Int. Ed. 2020, 59, 74–108.
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394.
- Le Vaillant, F.; Waser, J. Alkynylation of radicals: Spotlight on the “Third Way” to transfer triple bonds. Chem. Sci. 2019, 10, 8909–8923.
- Bietti, M. Activation and Deactivation Strategies Promoted by Medium Effects for Selective Aliphatic C–H Bond Functionalisation. Angew. Chem. Int. Ed. 2018, 57, 16618–16637.
- Galeotti, M.; Salamone, M.; Bietti, M. Electronic control over site-selectivity in hydrogen atom transfer (HAT) based C(sp3)–H functionalisation promoted by electrophilic reagents. Chem. Soc. Rev. 2022, 51, 2171–2223.
- Mayer, J.M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46.
- Lai, W.; Li, C.; Chen, H.; Shaik, S. Hydrogen-Abstraction Reactivity Patterns from A to Y: The Valence Bond Way. Angew. Chem. Int. Ed. 2012, 51, 5556–5578.
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685.
- Salamone, M.; Bietti, M. Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res. 2015, 48, 2895–2903.
- Zavitsas, A.A.; Pinto, J.A. Meaning of the polar effect in hydrogen abstractions by free radicals. Reactions of the tert-butoxy radical. J. Am. Chem. Soc. 1972, 94, 7390–7396.
- Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2002; pp. 18, 38, 41, 68, 69, 80, 146, 169, 170, 172, 209, 240–269, 291, 293, 298.
- Salamone, M.; Galeotti, M.; Romero-Montalvo, E.; van Santen, J.A.; Groff, B.D.; Mayer, J.M.; DiLabio, G.A.; Bietti, M. Bimodal Evans–Polanyi Relationships in Hydrogen Atom Transfer from C(sp3)–H Bonds to the Cumyloxyl Radical. A Combined Time-Resolved Kinetic and Computational Study. J. Am. Chem. Soc. 2021, 143, 11759–11776.
- Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693.
- Ide, T.; Barham, J.P.; Fujita, M.; Kawato, Y.; Egami, H.; Hamashima, Y. Regio- and chemoselective Csp3–H arylation of benzylamines by single electron transfer/hydrogen atom transfer synergistic catalysis. Chem. Sci. 2018, 9, 8453–8460.
- Choi, G.J.; Zhu, Q.; Miller, D.C.; Gu, C.J.; Knowles, R.R. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 2016, 539, 268–271.
- Roberts, B.P. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: Concepts and applications in organic chemistry. Chem. Soc. Rev. 1999, 28, 25–35.
- Giese, B. Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem. Int. Ed. 1983, 22, 753–764.
- Tedder, J.M. Which Factors Determine the Reactivity and Regioselectivity of Free Radical Substitution and Addition Reactions? Angew. Chem. Int. Ed. 1982, 21, 401–410.
- Ruffoni, A.; Mykura, R.C.; Bietti, M.; Leonori, D. The interplay of polar effects in controlling the selectivity of radical reactions. Nat. Synth. 2022, 1, 682–695.
- Chan, B.; Easton, C.J.; Radom, L. Outcome-Changing Effect of Polarity Reversal in Hydrogen-Atom-Abstraction Reactions. J. Phys. Chem. A 2015, 119, 3843–3847.
- Capaldo, L.; Ravelli, D.; Fagnoni, M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924.
- Galeotti, M.; Trasatti, C.; Sisti, S.; Salamone, M.; Bietti, M. Factors Governing Reactivity and Selectivity in Hydrogen Atom Transfer from C(sp3)–H Bonds of Nitrogen-Containing Heterocycles to the Cumyloxyl Radical. J. Org. Chem. 2022, 87, 7456–7463.
- Liu, F.; Ma, S.; Lu, Z.; Nangia, A.; Duan, M.; Yu, Y.; Xu, G.; Mei, Y.; Bietti, M.; Houk, K.N. Hydrogen Abstraction by Alkoxyl Radicals: Computational Studies of Thermodynamic and Polarity Effects on Reactivities and Selectivities. J. Am. Chem. Soc. 2022, 144, 6802–6812.
- Martin, T.; Galeotti, M.; Salamone, M.; Liu, F.; Yu, Y.; Duan, M.; Houk, K.N.; Bietti, M. Deciphering Reactivity and Selectivity Patterns in Aliphatic C–H Bond Oxygenation of Cyclopentane and Cyclohexane Derivatives. J. Org. Chem. 2021, 86, 9925–9937.
- Dantignana, V.; Milan, M.; Cussó, O.; Company, A.; Bietti, M.; Costas, M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358.
- Salamone, M.; Martin, T.; Milan, M.; Costas, M.; Bietti, M. Electronic and Torsional Effects on Hydrogen Atom Transfer from Aliphatic C–H Bonds: A Kinetic Evaluation via Reaction with the Cumyloxyl Radical. J. Org. Chem. 2017, 82, 13542–13549.
- Bietti, M.; Forcina, V.; Lanzalunga, O.; Lapi, A.; Martin, T.; Mazzonna, M.; Salamone, M. Kinetic Study of the Reaction of the Phthalimide-N-oxyl Radical with Amides: Structural and Medium Effects on the Hydrogen Atom Transfer Reactivity and Selectivity. J. Org. Chem. 2016, 81, 11924–11931.
- Salamone, M.; Mangiacapra, L.; Carboni, G.; Bietti, M. Hydrogen atom transfer from tertiary alkanamides to the cumyloxyl radical. The role of substrate structure on alkali and alkaline earth metal ion induced C–H bond deactivation. Tetrahedron 2016, 72, 7757–7763.
- Salamone, M.; Carboni, G.; Bietti, M. Fine Control over Site and Substrate Selectivity in Hydrogen Atom Transfer-Based Functionalisation of Aliphatic C–H Bonds. J. Org. Chem. 2016, 81, 9269–9278.
- Salamone, M.; DiLabio, G.A.; Bietti, M. Hydrogen Atom Abstraction Reactions from Tertiary Amines by Benzyloxyl and Cumyloxyl Radicals: Influence of Structure on the Rate-Determining Formation of a Hydrogen-Bonded Prereaction Complex. J. Org. Chem. 2011, 76, 6264–6270.
- Salamone, M.; Mangiacapra, L.; Bietti, M. Kinetic Solvent Effects on the Reactions of the Cumyloxyl Radical with Tertiary Amides. Control over the Hydrogen Atom Transfer Reactivity and Selectivity through Solvent Polarity and Hydrogen Bonding. J. Org. Chem. 2015, 80, 1149–1154.
- Salamone, M.; Carboni, G.; Mangiacapra, L.; Bietti, M. Binding to Redox-Inactive Alkali and Alkaline Earth Metal Ions Strongly Deactivates the C–H Bonds of Tertiary Amides toward Hydrogen Atom Transfer to Reactive Oxygen Centered Radicals. J. Org. Chem. 2015, 80, 9214–9223.
- Salamone, M.; Ortega, V.B.; Bietti, M. Enhanced Reactivity in Hydrogen Atom Transfer from Tertiary Sites of Cyclohexanes and Decalins via Strain Release: Equatorial C–H Activation vs Axial C–H Deactivation. J. Org. Chem. 2015, 80, 4710–4715.
- Salamone, M.; Basili, F.; Mele, R.; Cianfanelli, M.; Bietti, M. Reactions of the Cumyloxyl Radical with Secondary Amides. The Influence of Steric and Stereoelectronic Effects on the Hydrogen Atom Transfer Reactivity and Selectivity. Org. Lett. 2014, 16, 6444–6447.
- Milan, M.; Salamone, M.; Bietti, M. Hydrogen Atom Transfer from 1,n-Alkanediamines to the Cumyloxyl Radical. Modulating C–H Deactivation Through Acid–Base Interactions and Solvent Effects. J. Org. Chem. 2014, 79, 5710–5716.
- Kushch, O.V.; Hordieieva, I.O.; Kompanets, M.O.; Zosenko, O.O.; Opeida, I.A.; Shendrik, A.N. Hydrogen Atom Transfer from Benzyl Alcohols to N-Oxyl Radicals. Reactivity Parameters. J. Org. Chem. 2021, 86, 3792–3799.
- Paolillo, J.M.; Duke, A.D.; Gogarnoiu, E.S.; Wise, D.E.; Parasram, M. Anaerobic Hydroxylation of C(sp3)–H Bonds Enabled by the Synergistic Nature of Photoexcited Nitroarenes. J. Am. Chem. Soc. 2023, 145, 2794–2799.
- Ruffoni, A.; Hampton, C.; Simonetti, M.; Leonori, D. Photoexcited nitroarenes for the oxidative cleavage of alkenes. Nature. 2022, 610, 81–86.
- Hampton, C.; Simonetti, M.; Leonori, D. Olefin Dihydroxylation Using Nitroarenes as Photoresponsive Oxidants. Angew. Chem. Int. Ed. 2023, 62, e202214508.
- Gaster, E.; Kozuch, S.; Pappo, D. Selective Aerobic Oxidation of Methylarenes to Benzaldehydes Catalyzed by N-Hydroxyphthalimide and Cobalt(II) Acetate in Hexafluoropropan-2-ol. Angew. Chem. Int. Ed. 2017, 56, 5912–5915.
- Bietti, M.; Salamone, M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Role of Hydrogen Bonding. Org. Lett. 2010, 12, 3654–3657.
- Bietti, M.; Martella, R.; Salamone, M. Understanding Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. Org. Lett. 2011, 13, 6110–6113.
- Salamone, M.; Giammarioli, I.; Bietti, M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Importance of Solvent Hydrogen-Bond Interactions with the Substrate and the Abstracting Radical. J. Org. Chem. 2011, 76, 4645–4651.
- Paul, V.; Roberts, B.P. Homolytic reactions of ligated boranes. Part 8. Electron spin resonance studies of radicals derived from ligated alkylboranes. J. Chem. Soc. Perkin Trans. II 1988, 1183–1193.
- Paul, V.; Roberts, B.P.; Willis, C.R. Homolytic reactions of ligated boranes. Part 12. Amine–alkylboranes as polarity reversal catalysts for hydrogen-atom abstraction by t-butoxyl radicals. J. Chem. Soc. Perkin Trans. II 1989, 1953–1961.
- Horn, E.J.; Rosen, B.R.; Chen, Y.; Tang, J.; Chen, K.; Eastgate, M.D.; Baran, P.S. Scalable and sustainable electrochemical allylic C–H oxidation. Nature 2016, 533, 77–81.
- Sheldon, R.A. Synthesis and uses of alkyl hydroperoxides and dialkyl peroxides. In Peroxides; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1983; p. 169.
- Okada, K.; Okamoto, K.; Oda, M. A new and practical method of decarboxylation: Photosensitised decarboxylation of N-acyloxyphthalimides via electron-transfer mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738.
- Toriyama, F.; Cornella, J.; Wimmer, L.; Chen, T.G.; Dixon, D.D.; Creech, G.; Baran, P.S. Redox-Active Esters in Fe-Catalyzed C-C Coupling. J. Am. Chem. Soc. 2016, 138, 11132–11135.
- Cornella, J.; Edwards, J.T.; Qin, T.; Kawamura, S.; Wang, J.; Pan, C.-M.; Gianatassio, R.; Schmidt, M.; Eastgate, M.D.; Baran, P.S. Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc. 2016, 138, 2174–2177.
- Huihui, K.M.M.; Caputo, J.A.; Melchor, Z.; Olivares, A.M.; Spiewak, A.M.; Johnson, K.A.; DiBenedetto, T.A.; Kim, S.; Ackerman, L.K.G.; Weix, D.J. Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc. 2016, 138, 5016–5019.
- Leibler, I.N.-M.; Tekle-Smith, M.A.; Doyle, A.G. A general strategy for C(sp3)–H functionalisation with nucleophiles using methyl radical as a hydrogen atom abstractor. Nat. Commun. 2021, 12, 6950.
- Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.-H.; Chen, J.; Starr, J.T.; Baran, P.S. Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451.
- Cuthbertson, J.D.; MacMillan, D.W.C. The direct arylation of allylic sp3 C–H bonds via organic and photoredox catalysis. Nature. 2015, 519, 74–77.
- Madani, A.; Anghileri, L.; Heydenreich, M.; Möller, H.M.; Pieber, B. Benzylic Fluorination Induced by a Charge-Transfer Complex with a Solvent-Dependent Selectivity Switch. Org. Lett. 2022, 24, 5376–5380.
- Cismesia, M.A.; Yoon, T.P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434.
- Ye, T.; Zhang, F.-L.; Xia, H.-M.; Zhou, X.; Yu, Z.-X.; Wang, Y.-F. Stereoselective hydrogen atom transfer to acyclic radicals: A switch enabling diastereodivergent borylative radical cascades. Nat. Commun 2022, 13, 426.
- Katz, R.B.; Mistry, J.; Mitchell, M.B. An Improved Method for the Mono-Hydroxymethylation of Pyridines. A Modification of the Minisci Procedure. Synth. Commun. 1989, 19, 317–325.
- Chan, W.C.; Vinod, J.K.; Koide, K. Acetal Addition to Electron-Deficient Alkenes with Hydrogen Atom Transfer as a Radical Chain Propagation Step. J. Org. Chem. 2021, 86, 3674–3682.
- Curran, D.P.; Kim, D.; Liu, H.T.; Shen, W. Translocation of radical sites by intramolecular 1,5-hydrogen atom transfer. J. Am. Chem. Soc. 1988, 110, 5900–5902.
- Takahira, Y.; Chen, M.; Kawamata, Y.; Mykhailiuk, P.; Nakamura, H.; Peters, B.K.; Reisberg, S.H.; Li, C.; Chen, L.; Hoshikawa, T.; et al. Electrochemical C(sp3)–H Fluorination. Synlett 2019, 30, 1178–1182.
- Pulcinella, A.; Bonciolini, S.; Lukas, F.; Sorato, A.; Noël, T. Photocatalytic Alkylation of C(sp3)–H Bonds Using Sulfonylhydrazones. Angew. Chem. Int. Ed. 2023, 62, e202215374.
- Sarver, P.J.; Bacauanu, V.; Schultz, D.M.; DiRocco, D.A.; Lam, Y.-h.; Sherer, E.C.; MacMillan, D.W.C. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem. 2020, 12, 459–467.
- Proctor, R.S.J.; Phipps, R.J. Recent Advances in Minisci-Type Reactions. Angew. Chem. Int. Ed. 2019, 58, 13666–13699.
- Noble, A.; MacMillan, D.W.C. Photoredox α-Vinylation of α-Amino Acids and N-Aryl Amines. J. Am. Chem. Soc. 2014, 136, 11602–11605.
This entry is offline, you can click here to edit this entry!