Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The European Medicine Agency (EMA) has defined Adverse Drug Reactions (ADRs) as “a noxious and unintended response to a medicine”, not including poisoning, accidental, or intentional overdoses. The ADR occurrence differs based on the approach adopted for defining and detecting them, the characteristics of the population under study, and the research setting. ADRs have a significant impact on morbidity and mortality, particularly among older adults, and represent a financial burden for health services.
- adverse drug reactions
- older adults
- risk factors
1. Introduction
The International Conference of Harmonization (ICH) considers individuals aged 65 years or older as a “special population” because the differences with regard to comorbidity, polypharmacy, pharmacokinetics, and greater susceptibility to ADRs than the younger adults [1].
ADRs have a significant impact on morbidity and mortality, particularly among the elderly population, and are a financial burden for healthcare systems. It is estimated that between 30% and 60% of ADRs might be predictable and preventable. Indeed, they often arise from medication errors such as improper indications, high dosages, or prolonged treatment duration, as well as non-compliance with prescribed regimens or inappropriate self-medication in elderly and vulnerable patients. In addition to higher incidence, ADRs in older adults are more likely to be severe and underreported. Furthermore, the mortality rate associated with ADRs is significantly greater in older patients compared to younger individuals [2][3].
The care pathways for elderly patients can significantly differ from those of younger individuals with the same medical condition, particularly when considering treatment options and choices. The primary objective is not just a matter of treating a pathological condition, it is instead maintaining their independence, social engagement, and overall quality of life to the greatest extent possible. As a result, physicians are advised to consider patients’ health trajectories and needs to establish realistic therapeutic goals [4][5]. Following diagnosis, the process of medical prescription is mostly driven by the necessity to prevent the clinical manifestations and complications of the disease, including its interactions with concurrent conditions and pharmacological treatments. For these reasons, physicians should tailor the clinical recommendations outlined in the guidelines for a patient’s specific characteristics, rather than adhering strictly to disease-specific protocols. Another critical aspect involves the frequent reassessment of the ongoing treatment’s appropriateness. As older individuals often experience unstable health courses, the benefit/risk ratio of individual therapies may fluctuate in response to changes in their clinical conditions [4].
This strategy becomes even more important when one considers that drugs developed so far are not designed for individual patients, but for the average population; therefore, while working for the huge population majority, they are ineffective or even toxic for a part of it [6].
Several studies indicate that older adults may exhibit a higher incidence of ADRs compared to younger ones, and advancing age itself can be considered as a risk factor for ADR occurrence. In light of these findings, Stevenson et al. have proposed the necessity of adopting a comprehensive approach to address ADRs among the elderly population, treating drug-related harm as a geriatric syndrome [7].
As previously stated, numerous factors associated with ripe old age can lead to an elevated risk of incurring an ADR, such as higher rates of polypharmacy, multimorbidity, reduced organ function, frailty, age-related pharmacokinetic and pharmacodynamic variations, and individual variability in pharmacogenomics/pharmacogenetics. In addition, over the past few years, there has been growing recognition of the significant impact of genetic variations in drug-metabolizing and drug-transporting proteins on the occurrence of ADRs among older individuals. This is in line with the prevailing understanding that genomic variants can contribute to ADRs and can be effectively utilized to predict an individual’s response to drugs, encompassing both effectiveness and potential toxicity [8][9].
2. Polypharmacy
A significant age-related factor contributing to the higher prevalence of ADRs in the older population is represented by polypharmacy; indeed, multiple medications are commonly prescribed to manage various health issues simultaneously. International estimates indicate that over 60% of elderly individuals are prescribed five or more medications at the same time. The potential harm from drug reactions and interactions rises with the greater number of medicinal products, and the total number of drugs taken per day becomes a significant risk factor for ADR-related hospitalizations [10][11][12]. For instance, the risk of experiencing ADRs is 13% for a person on two medications, increasing to 58% and 82% for those taking five or seven or more medications per day, respectively [13]. These factors partially explain as such the elevated risk of adverse reactions observed in the older population, underlining the need for cautiousness when prescribing new drugs. Specific guidelines have been developed in Italy by Onder and colleagues with the aim of providing recommendations, currently lacking, for the care management of patients most susceptible to polypharmacy and multimorbidity. Such indications may not only improve the quality of individual care, but also assist the clinician, health professionals, and caregivers. The emphasis is on the need for multidimensional assessment through personalised and interdisciplinary approaches to identify patients most vulnerable to negative outcomes associated with polypharmacy. A limitation may be that there is inadequate evidence that the number of drugs per se, rather than inappropriate prescribing, is the direct cause of adverse consequences [4].
The majority of ADRs in the elderly belong to Type A, which are predictable and preventable with adequate evaluation and monitoring. Thus, prudent prescribing practices are essential in reducing errors and minimizing the risk of ADRs considering patient susceptibilities, medication history, and exploring non-pharmacological or conservative options [13][14][15].
Notably, the eldest and frail individuals are frequently excluded from clinical trials, making it challenging to hypothesise the nature and incidence of adverse events. Moreover, guidelines predominantly focus on managing single diseases, so strict adherence to them when prescribing may be detrimental when dealing with older individuals with multiple comorbidities [16].
3. Multimorbidity
Multimorbidity refers to the coexistence of at least two chronic diseases in the same patient. Particularly in geriatrics, multimorbidity is a significant concern as it is closely linked to the occurrence of iatrogenic illnesses. Several studies have highlighted that the risk of ADRs escalates with an increasing number of chronic diseases. This can be attributed to various factors, including a higher likelihood of drug–disease interactions, as medications used for one condition may worsen symptoms of other underlying disorders [17][18][19].
4. Changes in Drug Metabolism
The aging process significantly affects the body’s homeostasis, in one with physiological processes, that, possibly, increase the risk of iatrogenic events [20], therefore pharmacokinetic alterations due to age, as well as factors like multimorbidity, frailty, and polypharmacy, as mentioned above, may play a critical role in this circumstance [15][21][22]. Changes in pharmacokinetics impact drug metabolism and clearance, thereby heightening the risk of ADRs, or altered drug responsiveness [23]. Modifications in the distribution volumes of drugs, attributable to a lower body fluid content and differences in lipid distribution, can contribute to the prolongation of the half-life of a given drug, thus escalating the probability of toxicity. In patients on polypharmacy, drug metabolism can also be affected by interactions with CYP450 enzymes. A cross-sectional study of eighty-year-old institutional and community residents revealed that 72.2% of individuals exhibited potential CYP drug-to-drug interactions that affected not only their functional performance and mobility, but also their self-perception of health [20][24]. So far, the impact of gender on the incidence of ADRs has been clearly demonstrated in a large number of studies. In particular, a systematic analysis aimed at assessing the extent of sex differences in ADRs across a wide range of treatments has found that slightly less than half of the medicines investigated (307 vs. 668) show a different sex-related rate of ADRs [25].
Gender-based differences in genetics, immunology, pharmacokinetics, and pharmacodynamic may account for the existing variability of ADRs, with women being generally more susceptible than men [26].
In addition, levels of sex steroid hormones, which contribute to differences in drug response, have been shown to change with age, thus potentially directly and indirectly affecting drugs’ ADME. Understanding physiological variations between the two sexes would enable personalised medicine to move forward [27][28].
5. Geriatric Syndromes
Geriatric syndromes encompass a range of conditions such as falls, delirium, cognitive impairment, orthostatic hypotension, incontinence, and chronic pain, which can reduce the potential benefits of pharmacological treatments [29][30], increase the risk of ADRs, and contribute to inappropriate prescriptions [31]. For instance, older adults taking oral antidiabetic medications face a heightened vulnerability to hypoglycaemia, thereby raising the risk of falls. Certain medications such as anticonvulsants, antidepressants, and some anti-Parkinsons drugs have been linked to an elevated risk of delirium and incontinence. Treatments for chronic pain, such as opioid agonists, have also been associated with delirium and an increased risk of falls. Moreover, some treatments may have indirect fatal consequences, as in patients with atrial fibrillation who are at high risk of falls, as anticoagulant therapy has been shown to elevate the risk of intracranial bleeding [32].
6. Pharmacogenetics/Genomics Variability
Pharmacogenetics/genomics is an evolving field with significant potential regarding its clinical application in tailoring therapy to optimize effectiveness and minimize the risk of adverse reactions. Pharmacogenetics focuses on the study of genetic factors that contribute to individual variations in drug response, while pharmacogenomics involves the genome-wide analysis of genetic determinants of drug efficacy and toxicity. The main distinction between pharmacogenetics and pharmacogenomics lies in the former’s emphasis on examining a few specific genes, whereas the latter encompasses the study of genes across all chromosomes [33][34][35]. The advancements in this field can be largely attributed to the increasing availability of information about the human genome, which has been made accessible to the entire scientific community since the completion of the human genome project in 2003. Thanks to progresses in bioinformatics, the massive volume of data derived from human genome sequencing has been organized, processed, and catalogued in databases [36].
A pharmacogenetic test must meet certain necessary criteria to be used in clinical practice. First, an established association between a genotype and the response to a specific drug is needed, either in the general population or a specific subgroup. The test must demonstrate both clinical and analytical validity. The method used to determine the genotype should exhibit sufficient sensitivity, specificity, and predictive values. Lastly, there should be scientific evidences supporting the utility of the test, meaning its application in a clinical setting should enhance treatment response, patient compliance, and ultimately improve the benefit/risk ratio associated with drug administration [37].
The majority of phase I reactions, such as oxidation, reduction, and hydrolysis, involved in drug metabolism to activate a prodrug or convert parent drugs to active or inactive metabolites, are facilitated by the cytochrome P450 superfamily of haemoproteins [38]. Among those, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 are considered the most significant for drug metabolism. They are responsible for approximately 90% of the systemic clearance and bioavailability of human drugs [39]. Medicinal products interacting with the CYP450 isoenzymes can be classified as substrates, inhibitors, or inducers. Inhibitors can be further characterized as being weak, moderate or potent [40].
Functional CYP polymorphisms encompass a wide range of genetic variations, including gene deletions and duplications, frame shift mutations, amino acid changes, intronic mutations (leading to altered splicing sites), and copy number variations in functional gene copies. Generally, the population can be classified into three major phenotypes based on a specific CYP450 enzyme: ultrarapid metabolizers, who possess more than two active genes encoding a particular P450 enzyme; extensive metabolizers, who carry two functional genes; poor metabolizers, lacking functional enzymes due to defective or deleted genes. Additionally, an intermediate metabolizer phenotype is often considered, encompassing individuals with either one functional and one defective allele, or two partially defective alleles [9][41][42].
Hence, variation in genes encoding drug-metabolising enzymes, drug transporters, and drug targets affects drug disposition and action, contributing to variability in drug response and in the development of ADRs. Thus, it may be useful in clinical practice to recognise the importance of genetic variation in contributing to the potential occurrence of ADRs, given the large number of genes involved in drug metabolism and transport. However, patients, especially elderly, may receive multiple inhibitors of a given cytochrome or an inhibitor and an inducer of the same one, making it difficult to predict the clinical relevance of these interactions. Furthermore, it is not enough to identify a clinically significant interaction; it would also be necessary to inform patients, modify the treatment plan, and perform regular follow-up to assess the safety and efficacy of the new pharmacotherapy. Nonetheless, in light of recent advances in pharmacogenetics/genomics, it would be desirable to revise the conventional understanding of DDIs encompassing the influence of genetic variations. Indeed, by applying pharmacogenetics/genomics a profile of a patient’s gene variations may be created, prior to administration of a drug. To date, a single open label, multicentre, controlled, cluster-randomized, crossover implementation study investigating the beneficial impact of pharmacogenomics testing, has been performed. Despite its clinical utility not yet being established, encouraging results have emerged showing that clinically relevant ADRs reduced by about 30% with pharmacogenetics-guided prescribing [43].
A summary of risk factors involved in ADRs occurrence is provided in Figure 1.
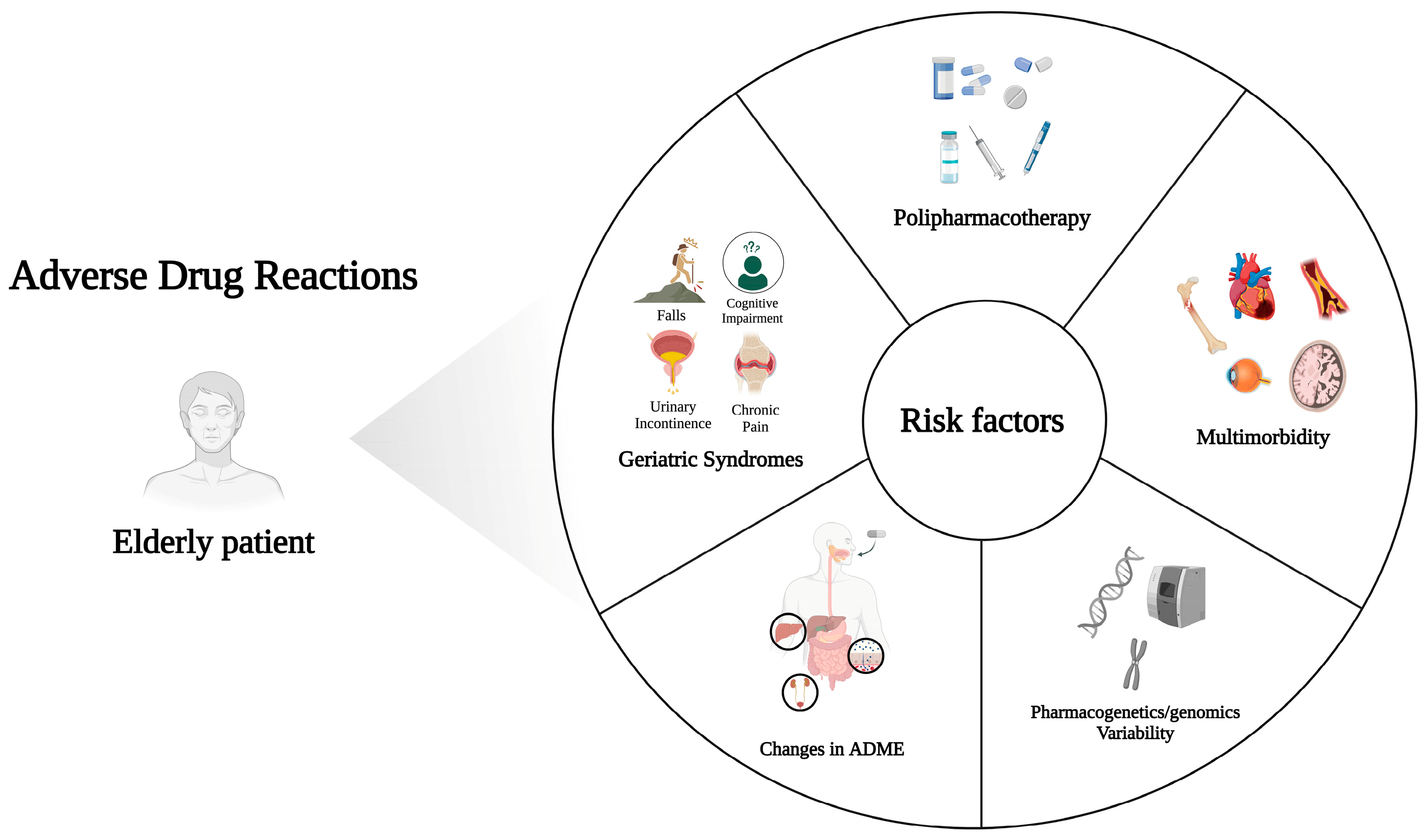
Figure 1. Summary of risk factors leading to ADRs in elderly patients. Created with BioRender.com; accessed on 19 October 2023.
6.1. Drug–Drug Interactions (DDIs), Drug–Gene Interactions (DGIs), and Drug–Drug–Gene Interactions (DDGIs)
Drug–drug interactions (DDIs) can be divided into pharmacodynamic (PD) and pharmacokinetic (PK) interactions. PD interactions occur when drugs cause additive or antagonistic pharmacological effects that influence safety and/or efficacy. The co-administration of warfarin and NSAIDs is an example of a PD interaction, as their concomitant use may increase the risk of bleeding. PK interactions may be due to changes in absorption, distribution (protein and tissue binding), metabolism, and excretion [44].
In terms of PK, drug–gene interactions (DGIs), occur when an individual carrying one or more variant forms of a gene encoding a drug metabolizing enzyme or drug transporter with altered function receives a drug that is a substrate for the given enzyme or transporter [45].
Drug–drug–gene interactions (DDGIs) arise when both drugs and an individuals’ genetic profile alter the efficacy and/or safety of a specified medicinal product [46]; this notion will help to understand the phenoconversion interactions described below.
It is worth noting that in the real-world setting the interpretation of metabolic phenotypes should be evaluated considering DDGIs as a consequence of polypharmacy, especially in elderly. Yet the majority of pharmacogenetic recommendations are still based on the more well documented single gene–drug interaction, although the concomitant administration of another medication could influence the individual response [47].
Interaction mechanisms can be divided into three main categories: induction, inhibitory, and phenoconversion. The first two refer to any interactions that may impact PK of the victim drug, either by increasing or decreasing its concentrations. The latter can arise from the administration of a perpetrator drug that affects the metabolism or transport of the victim drug, as well as the presence of genetic variants leading to loss- or gain-of-function (LOF or GOF) in enzymes responsible for the metabolism or transport of the victim drug, or even a combination of both factors.
A phenoconversion can occur when the combined effect of the interacting drug and the genotype produces opposing effects, resulting in a temporary shift in phenotype [48].
As the management of DDIs largely depends on the clinical impact and severity of the interaction, many tools are available to determine their clinical significance. However, there is poor agreement among the current resources and a standardised classification method would be warranted. More specifically, the British National Formulary marks with bullet points potentially harmful drug pairs which should be prescribed cautiously, under appropriate monitoring, or avoided altogether [49]. The Micromedex Drug–Reax System categorises interactions into three degrees of severity, major, moderate, and minor, and the strength of the reporting into five categories—excellent, good, fair, poor, and unlikely [50]. The Drugs.com Drug Interaction Checker (DDIC) classifies interactions into four severity levels: major, moderate, minor, and unknown [51]. Vidal’s Interactions médicamenteuses comprises four seriousness grades according to the recommended clinical management—contraindicated, avoid, precaution, and “take into account” (i.e., no specific recommendation) [52]. Drug Interaction Facts rates interaction severity into three levels—major, moderate, and minor—and the degree of documentation into five—established, probable, suspected, possible, and unlikely—by combining these two categories. It also ranks each interaction from 1 to 5 in terms of importance [53].
Induction, Inhibitory, and Phenoconversion Interactions
Increased metabolism of active drugs due to the presence of an enzyme inducer or GOF variant can lead to a decrease in the effectiveness of the substrate drug. Prodrugs exhibit an opposite effect. When an enzyme-inducing drug or GOF variant are involved in their metabolism, elevated plasma levels of active metabolites may occur, leading to increased side effects and/or efficacy. For instance, individuals with CYP2C19*17 GOF variants experience enhanced conversion of clopidogrel to active metabolites, which in turn reduce the occurrence of cardiovascular events, but may increase the likelihood of bleeding episodes, which would be particularly risky in older and frail adults. Indeed, a five-year review of spontaneous pharmacovigilance reports elucidated an increased susceptibility to ADRs related to antithrombotic medicinal products [54]. Furthermore, Dubrall et al. emphasise the need to continuously monitor the prescription of antithrombotics in the elderly, as they account for a high proportion of ADRs in this cohort of patients [55]. If a CYP1A2, CYP2C9, and/or CYP3A4 inducer is co-administered, this is expected to enhance the efficacy of clopidogrel, but also an increase in bleeding risk [56][57][58][59][60].
The inhibitory effects of drugs and genotype can influence substrate metabolism by affecting either the same metabolizing enzyme or different metabolic pathways through drug and genotype interactions. Generally, individuals classified as poor metabolizers are expected to have the highest plasma concentration of substrate drug when co-administered with inhibitor, compared to other metabolic genotypes. For instance, the co-administration of simvastatin, a CYP2C9 inhibitor, with warfarin, a CYP2C9 substrate, has demonstrated a reduction in warfarin dosage requirements, particularly in CYP2C9*3 carriers, with a significantly higher percentage compared to noncarriers (29% vs. 5%, respectively) [61]. Nevertheless, the inhibitory effects of drugs and genotypes do not always exhibit additive behaviour. Genetically poor metabolizers are likely to experience only limited additional enzyme inhibition when an inhibitory drug is administered. For example, a statistically significant increase in rabeprazole, a CYP2C19 substrate, in plasma levels were observed in both normal metabolizers and carriers of the heterozygous genotype after treatment with fluvoxamine (CYP2C19 inhibitor). Nonetheless, no further clinically meaningful increase was detected in poor metabolizers, who already displayed highest rabeprazole plasma levels [62].
If a drug is metabolized by multiple CYP enzymes, inhibiting only one of those (via drug or genotype) may have minimal impact, due to the presence of alternative pathways. However, when both the genotype and the interacting drug affect different routes of metabolism, interactions may be significant.
A large proportion of prodrugs require specific CYP enzymes to become therapeutically active, taking clopidogrel as an example, which relies on the activation of CYP1A2, CYP2B6, CYP3A4, CYP2C9, and CYP2C19 [63]; individuals carrying LOF variants in one or more of these genes, and who are co-treated with inhibitors of these enzymes, face an increased risk of treatment resistance. For example, carriers of CYP2C19*2 and/or *3 alleles who receive clopidogrel alongside proton pump inhibitors, CYP2C19 inhibitors, are more prone to experience reduced efficacy of clopidogrel. Additionally, the introduction of a third risk factor, such as calcium channel blockers, CYP3A4 inhibitors, is associated with an even greater reduction in clopidogrel effectiveness [64][65].
A temporary change in phenotype can occur when the effect of a perpetrator drug is opposed to the genetic one. For instance, individuals with reduced function CYP2C9 variants exhibit decreased metabolism of tolbutamide, a CYP2C9 substrate. However, co-administration of rifampicin, an inducer of CYP2C9, in these patients counteracts the genetic effect, leading to a twofold increase in tolbutamide clearance [66]. On the other hand, when proton pump inhibitors are used alongside clopidogrel, a phenoconversion occurs, transforming individuals with genetically determined ultra-rapid metabolism into poor metabolizers, as evidenced by the loss of clopidogrel’s efficacy [67].
The bright side of phenoconversion interactions is that genetically determined phenotypes can be restored to normal by introducing medications with opposing effects on metabolism. As an example, resistance to nortriptyline, a CYP2D6 substrate, caused by excessively rapid metabolism, can be successfully reversed and normalized by adding paroxetine a CYP2D6 inhibitor, resulting in a restoration of therapeutic plasma levels of nortriptyline [68].
6.2. Drug–Drug-Transporters–Genes Interactions (DDTGIs)
Drug transporters regulate the “movement” of pharmaceutical compounds to and from various body districts. Key locations where these transporters impact drug pharmacokinetics include liver, kidneys, blood–brain barrier (BBB), and intestine.
Transporters can be classified into two main categories, namely efflux transporters (divided into group I and group II accordingly to transport direction) and uptake transporters (group III).
Similar to the drug metabolizing enzyme scenario, these interactions may be intensified or reversed, via inhibitory/induction or phenoconversion pathways [69]. Nonetheless, some issues need to be considered in respect to drug–drug-transporters interactions in clinical practice. Indeed, there are a very limited number of drugs, if any, whose carrying depends on a single transporter; the victim drug may be a substrate of other uptake or efflux transporters. Furthermore, a large proportion of victim drugs are not only substrates of one or more transporters, but also metabolised by one or more phase I and/or phase II enzymes. Perpetrator drugs are also often not specific for a single transporter, but they may inhibit or induce other transporters and/or drug metabolising enzymes. Thus, it is still difficult to obtain clear data on in vivo contributions to overall changes in the pharmacokinetics of victim drugs for each specific transporter-mediated drug–drug interactions. Additionally, during drug development, not all theoretically possible drug combinations can be tested for transporter-mediated drug–drug interactions, nor it is common practice to test the effect of all known inhibitors on the transport of a target drug. Extrapolation of inhibition data obtained with classical transporter inhibitors and translation to other inhibitors of the same transporter is challenging (e.g., due to different binding sites at the transporter), in particular in elderly patients who, as written above, receive multiple drugs.
P-glycoprotein 1 (P-gp, ABCB1), multidrug resistance-associated protein 2 (MRP2, ABCC2), and breast cancer resistant protein (BCRP, ABCG2) transporters are present in the bowel, liver, kidney, and blood–brain barrier (BBB). These transporters play a role in effluxing substrates back into the intestinal lumen, facilitating hepatic and renal excretion (except for BCRP), and functioning inversely at the BBB to protect the brain from foreign substances entry and to redirect the latter into the systemic circulation. Inhibition of their function in the intestine, liver, or kidney can lead to increased systemic exposure of substrates (although the opposite effect is expected when inhibiting transport across the BBB).
In the liver, kidney, and BBB, essential uptake transporters, such as organic cation transporters (OCTs) 1/2/3, organic anion-transporting polypeptides (OATPs) 1B1/1B3/2B1, and multidrug and toxic compound extrusion proteins (MATEs) 1/2, all follow a common route for transporting their substrates from the blood stream into various tissues, or into urine/bile. Consequently, modulating the capacities of these transporters would determine increased or decreased systemic drug concentrations. However, a contrary effect can be observed with the uptake transporters expressed in the apical membrane of the intestine, such as OATPs and OCT1, as the transportation pathway occurs in the opposite direction. In certain cases, altering the function of uptake transporters can increase the risk of ADRs. For instance, individuals with two reduced function alleles of OCT1 (SLC22A1) treated with OCT1 inhibitors were more than four times likely to experience gastrointestinal side effects during metformin (an OCT1 substrate) therapy, attributable to the accumulation of metformin in the intestinal lumen. This finding is supported by a previous study as well [70][71].
At the level of renal uptake transporters, other drug–drug-transporter gene interactions have been reported, where carrying mutant alleles and co-administration of inhibitors were associated with increased plasma levels/toxicity or reduced clearance of metformin [72][73]. On the other hand, reducing transport may decrease specific side effects. For instance, individuals carrying the rs316019 (C > A) mutation in OCT2 (SLC22A2) were protected against nephrotoxicity and ototoxicity caused by cisplatin (OCT2 substrate). The variant resulted in reduced transport of cisplatin into the kidneys and the inner ear (cochlea) where OCT2 is also expressed [74][75].
In many cases, the effectiveness of a drug depends on its ability to access certain tissues. Statins, for example, enter the liver by OATP1B1 (SLCO1B1), which is crucial for their lipid-lowering effects. Reducing this uptake pathway diminishes statin efficacy, leading to elevated plasma concentrations, and results in myopathy and, in rare occasions, rhabdomyolysis. The rs4149056 (T > C) variant (SLCO1B1*15) has been extensively studied and consistently associated with increased statin plasma exposure, muscle aches, dose reduction, and/or treatment-resistant phenotypes [76][77][78][79].
7. Strategies Supporting the Appropriateness of Drug Use and Prevention of ADRs in Elderly Patients
Refining drug therapy is an essential aspect of caring for elderly patients. The process of prescribing a medication includes specific information to be considered, such as drug indication, determining a dose, monitoring for effectiveness and toxicity, educating the patient about expected side effects, and indications for seeking consultation. Therefore, ADRs are the serious consequences of inappropriate drug prescribing [80].
As highlighted above, polypharmacy is a major risk factor for ADRs’ onset. Therefore, one of the most important interventions to reduce the threat of iatrogenic disease is to decrease the burden of medicines.
Deprescribing refers to the supervised process of discontinuing inappropriate medications or reducing their dosage enhancing patient outcomes [81][82][83]. Scott et al. propose a five-step protocol to facilitate this procedure. These steps involve conducting a review of the patient’s medications to assess their appropriateness in relation to clinical condition, overall functioning, life expectancy, and health priorities. Based on this evaluation, each drug should be carefully examined, bearing in mind the danger of ADRs and the benefit/risk ratio. Once the drug to be discontinued is identified, it is crucial to monitor for potential withdrawal reactions or improvements in outcomes [84].
Prescribing and deprescribing processes require meticulous documentation of the patient’s health status. This involves identifying clinical geriatric conditions, conducting a comprehensive review of medications (including herbal remedies and over-the-counter drugs), carefully analysing any previous ADRs, and establishing clear health priorities and treatment objectives. In older individuals with polypharmacotherapy, the introduction of new medicines should involve gradual titration to minimize the risk of adverse events, and any new symptom should be evaluated as potential ADRs [13]. This appears critical to prevent the occurrence of a prescribing cascade, which transpires when an additional medicine is prescribed to counteract an ADR that is mistakenly interpreted as a new medical condition. A classic example of this sequence is the prescription of anti-Parkinson drugs to manage motor symptoms associated with prolonged antipsychotic therapy [85].
In this scenario, adherence to therapy plays a key role. After determining the most suitable therapeutic approach, physicians should devote sufficient efforts and time to inform and engage patients, as well as their caregivers (e.g., family members or non-healthcare professionals responsible for the well-being of an elderly or dependent individual), and other healthcare professionals involved in their care. Effective physician–patient interaction is essential for enhancing the patient’s understanding of medical recommendations and promoting acceptance of the prescribed treatments [86].
Several studies have demonstrated that open communication between physicians and patients regarding diagnosis and treatment, thus incorporating shared decision making, improves adherence to medical advice and achieves positive short- and medium-term clinical outcomes [87][88][89]. In addition to ensuring that patients and caregivers are aware about necessity, role, and potential adverse effects of the prescription, acceptance may be influenced by various factors following treatment initiation. These determinants include the therapeutic benefits in terms of disease control and quality of life, the tolerability of medicines, and the convenience of drug administration according to formulation and dosage [90][91].
A facilitator of medication adherence is the establishment of interpersonal trust between physician and patient, essential in the patient–physician relationship, particularly among older patients. Research by Thom et al. has shown that low trust in physicians is associated with poorer adherence to medical recommendations, reduced satisfaction with care, and limited improvement in symptoms. Furthermore, when patients relay on their medical doctors, they are more likely to disclose their health-related behaviours, even those they may consider embarrassing or sensitive [92][93][94].
This entry is adapted from the peer-reviewed paper 10.3390/ph16111542
References
- EMA. ICH E7 Studies in Support of Special Populations: Geriatrics-Scientific Guideline. Available online: https://www.ema.europa.eu/en/ich-e7-studies-support-special-populations-geriatrics-scientific-guideline (accessed on 28 September 2023).
- McLean, A.J.; Le Couteur, D.G. Aging Biology and Geriatric Clinical Pharmacology. Pharmacol. Rev. 2004, 56, 163–184.
- Atkin, P.A.; Veitch, P.C.; Veitch, E.M.; Ogle, S.J. The Epidemiology of Serious Adverse Drug Reactions among the Elderly. Drugs Aging 1999, 14, 141–152.
- Onder, G.; Vetrano, D.L.; Palmer, K.; Trevisan, C.; Amato, L.; Berti, F.; Campomori, A.; Catalano, L.; Corsonello, A.; Kruger, P.; et al. Italian Guidelines on Management of Persons with Multimorbidity and Polypharmacy. Aging Clin. Exp. Res. 2022, 34, 989–996.
- Muth, C.; Blom, J.W.; Smith, S.M.; Johnell, K.; Gonzalez-Gonzalez, A.I.; Nguyen, T.S.; Brueckle, M.-S.; Cesari, M.; Tinetti, M.E.; Valderas, J.M. Evidence Supporting the Best Clinical Management of Patients with Multimorbidity and Polypharmacy: A Systematic Guideline Review and Expert Consensus. J. Intern. Med. 2019, 285, 272–288.
- Obreli Neto, P.R.; Nobili, A.; de Lyra, D.P.; Pilger, D.; Guidoni, C.M.; de Oliveira Baldoni, A.; Cruciol-Souza, J.M.; de Carvalho Freitas, A.L.; Tettamanti, M.; Gaeti, W.P.; et al. Incidence and Predictors of Adverse Drug Reactions Caused by Drug-Drug Interactions in Elderly Outpatients: A Prospective Cohort Study. J. Pharm. Pharm. Sci. 2012, 15, 332–343.
- Stevenson, J.M.; Davies, J.G.; Martin, F.C. Medication-Related Harm: A Geriatric Syndrome. Age Ageing 2019, 49, 7–11.
- Conforti, A.; Costantini, D.; Zanetti, F.; Moretti, U.; Grezzana, M.; Leone, R. Adverse Drug Reactions in Older Patients: An Italian Observational Prospective Hospital Study. Drug Healthc. Patient Saf. 2012, 4, 75–80.
- Cardelli, M.; Marchegiani, F.; Corsonello, A.; Lattanzio, F.; Provinciali, M. A Review of Pharmacogenetics of Adverse Drug Reactions in Elderly People. Drug Saf. 2012, 35 (Suppl. S1), 3–20.
- Gutiérrez-Valencia, M.; Izquierdo, M.; Cesari, M.; Casas-Herrero, Á.; Inzitari, M.; Martínez-Velilla, N. The Relationship between Frailty and Polypharmacy in Older People: A Systematic Review. Br. J. Clin. Pharmacol. 2018, 84, 1432–1444.
- Duerden, M.; Avery, T.; Payne, R. Polypharmacy and Medicines Optimisation: Making It Safe and Sound; The King’s Fund: London, UK, 2013.
- Onder, G.; Pedone, C.; Landi, F.; Cesari, M.; Della Vedova, C.; Bernabei, R.; Gambassi, G. Adverse Drug Reactions as Cause of Hospital Admissions: Results from the Italian Group of Pharmacoepidemiology in the Elderly (GIFA). J. Am. Geriatr. Soc. 2002, 50, 1962–1968.
- Davies, E.A.; O’Mahony, M.S. Adverse Drug Reactions in Special Populations—The Elderly. Br. J. Clin. Pharmacol. 2015, 80, 796–807.
- Coleman, J.J.; Pontefract, S.K. Adverse Drug Reactions. Clin. Med. 2016, 16, 481–485.
- Ventura, M.T.; Laddaga, R.; Cavallera, P.; Pugliese, P.; Tummolo, R.A.; Buquicchio, R.; Pierucci, P.; Passalacqua, G. Adverse Drug Reactions as the Cause of Emergency Department Admission: Focus on the Elderly. Immunopharmacol. Immunotoxicol. 2010, 32, 426–429.
- Routledge, P.A.; O’Mahony, M.S.; Woodhouse, K.W. Adverse Drug Reactions in Elderly Patients. Br. J. Clin. Pharmacol. 2004, 57, 121–126.
- Lattanzio, F.; Landi, F.; Bustacchini, S.; Abbatecola, A.M.; Corica, F.; Pranno, L.; Corsonello, A. Geriatric Conditions and the Risk of Adverse Drug Reactions in Older Adults: A Review. Drug Saf. 2012, 35 (Suppl. S1), 55–61.
- Calderón-Larrañaga, A.; Vetrano, D.L.; Onder, G.; Gimeno-Feliu, L.A.; Coscollar-Santaliestra, C.; Carfí, A.; Pisciotta, M.S.; Angleman, S.; Melis, R.J.F.; Santoni, G.; et al. Assessing and Measuring Chronic Multimorbidity in the Older Population: A Proposal for Its Operationalization. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1417–1423.
- Onder, G.; Lattanzio, F.; Battaglia, M.; Cerullo, F.; Sportiello, R.; Bernabei, R.; Landi, F. The Risk of Adverse Drug Reactions in Older Patients: Beyond Drug Metabolism. Curr. Drug Metab. 2011, 12, 647–651.
- Mangoni, A.A.; Jackson, S.H.D. Age-Related Changes in Pharmacokinetics and Pharmacodynamics: Basic Principles and Practical Applications. Br. J. Clin. Pharmacol. 2004, 57, 6–14.
- Budnitz, D.S.; Pollock, D.A.; Weidenbach, K.N.; Mendelsohn, A.B.; Schroeder, T.J.; Annest, J.L. National Surveillance of Emergency Department Visits for Outpatient Adverse Drug Events. JAMA 2006, 296, 1858–1866.
- Budnitz, D.S.; Lovegrove, M.C.; Shehab, N.; Richards, C.L. Emergency Hospitalizations for Adverse Drug Events in Older Americans. N. Engl. J. Med. 2011, 365, 2002–2012.
- Klotz, U. Pharmacokinetics and Drug Metabolism in the Elderly. Drug Metab. Rev. 2009, 41, 67–76.
- Verde, Z.; García de Diego, L.; Chicharro, L.M.; Bandrés, F.; Velasco, V.; Mingo, T.; Fernández-Araque, A. Physical Performance and Quality of Life in Older Adults: Is There Any Association between Them and Potential Drug Interactions in Polymedicated Octogenarians. Int. J. Environ. Res. Public Health 2019, 16, 4190.
- Yu, Y.; Chen, J.; Li, D.; Wang, L.; Wang, W.; Liu, H. Systematic Analysis of Adverse Event Reports for Sex Differences in Adverse Drug Events. Sci. Rep. 2016, 6, 24955.
- Moyer, A.M.; Matey, E.T.; Miller, V.M. Individualized Medicine: Sex, Hormones, Genetics, and Adverse Drug Reactions. Pharmacol. Res. Perspect. 2019, 7, e00541.
- Tran, C.; Knowles, S.R.; Liu, B.A.; Shear, N.H. Gender Differences in Adverse Drug Reactions. J. Clin. Pharmacol. 1998, 38, 1003–1009.
- Franconi, F.; Brunelleschi, S.; Steardo, L.; Cuomo, V. Gender Differences in Drug Responses. Pharmacol. Res. 2007, 55, 81–95.
- Saraf, A.A.; Petersen, A.W.; Simmons, S.F.; Schnelle, J.F.; Bell, S.P.; Kripalani, S.; Myers, A.P.; Mixon, A.S.; Long, E.A.; Jacobsen, J.M.L.; et al. Medications Associated with Geriatric Syndromes and Their Prevalence in Older Hospitalized Adults Discharged to Skilled Nursing Facilities. J. Hosp. Med. 2016, 11, 694–700.
- Vetrano, D.L.; Palmer, K.; Marengoni, A.; Marzetti, E.; Lattanzio, F.; Roller-Wirnsberger, R.; Lopez Samaniego, L.; Rodríguez-Mañas, L.; Bernabei, R.; Onder, G.; et al. Frailty and Multimorbidity: A Systematic Review and Meta-Analysis. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 659–666.
- Muhlack, D.C.; Hoppe, L.K.; Stock, C.; Haefeli, W.E.; Brenner, H.; Schöttker, B. The Associations of Geriatric Syndromes and Other Patient Characteristics with the Current and Future Use of Potentially Inappropriate Medications in a Large Cohort Study. Eur. J. Clin. Pharmacol. 2018, 74, 1633–1644.
- Gage, B.F.; Birman-Deych, E.; Kerzner, R.; Radford, M.J.; Nilasena, D.S.; Rich, M.W. Incidence of Intracranial Hemorrhage in Patients with Atrial Fibrillation Who Are Prone to Fall. Am. J. Med. 2005, 118, 612–617.
- Food and Drug Administration, HHS. International Conference on Harmonisation. Guidance on E15 Pharmacogenomics Definitions and Sample Coding; Availability. Notice. Fed. Regist. 2008, 73, 19074–19076.
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating Functional Genomics into Rational Therapeutics. Science 1999, 286, 487–491.
- PharmGKB. Available online: https://www.pharmgkb.org/ (accessed on 6 July 2023).
- Pirazzoli, A.; Recchia, G. Pharmacogenetics and Pharmacogenomics: Are They Still Promising? Pharmacol. Res. 2004, 49, 357–361.
- Lee, K.C.; Ma, J.D.; Kuo, G.M. Pharmacogenomics: Bridging the Gap between Science and Practice. J. Am. Pharm. Assoc. 2010, 50, e1–e17.
- Pinto, N.; Dolan, M.E. Clinically Relevant Genetic Variations in Drug Metabolizing Enzymes. Curr. Drug Metab. 2011, 12, 487–497.
- Singh, D.; Kashyap, A.; Pandey, R.V.; Saini, K.S. Novel Advances in Cytochrome P450 Research. Drug Discov. Today 2011, 16, 793–799.
- Huang, S.-M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J.; et al. New Era in Drug Interaction Evaluation: US Food and Drug Administration Update on CYP Enzymes, Transporters, and the Guidance Process. J. Clin. Pharmacol. 2008, 48, 662–670.
- Nelson, D.R.; Zeldin, D.C.; Hoffman, S.M.G.; Maltais, L.J.; Wain, H.M.; Nebert, D.W. Comparison of Cytochrome P450 (CYP) Genes from the Mouse and Human Genomes, Including Nomenclature Recommendations for Genes, Pseudogenes and Alternative-Splice Variants. Pharmacogenetics 2004, 14, 1–18.
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of Cytochrome P450 Polymorphisms on Drug Therapies: Pharmacogenetic, Pharmacoepigenetic and Clinical Aspects. Pharmacol Ther. 2007, 116, 496–526.
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.-C.; et al. A 12-Gene Pharmacogenetic Panel to Prevent Adverse Drug Reactions: An Open-Label, Multicentre, Controlled, Cluster-Randomised Crossover Implementation Study. Lancet 2023, 401, 347–356.
- Shorr, R.I.; Ray, W.A.; Daugherty, J.R.; Griffin, M.R. Concurrent Use of Nonsteroidal Anti-Inflammatory Drugs and Oral Anticoagulants Places Elderly Persons at High Risk for Hemorrhagic Peptic Ulcer Disease. Arch. Intern. Med. 1993, 153, 1665–1670.
- Westervelt, P.; Cho, K.; Bright, D.R.; Kisor, D.F. Drug-Gene Interactions: Inherent Variability in Drug Maintenance Dose Requirements. Pharm. Ther. 2014, 39, 630–637.
- Hahn, M.; Roll, S.C. The Influence of Pharmacogenetics on the Clinical Relevance of Pharmacokinetic Drug-Drug Interactions: Drug-Gene, Drug-Gene-Gene and Drug-Drug-Gene Interactions. Pharmaceuticals 2021, 14, 487.
- Peñas-LLedó, E.; LLerena, A. Clinical Use of Pre-Emptive Pharmacogenetic Programmes. Lancet 2023, 401, 320–321.
- Malki, M.A.; Pearson, E.R. Drug–Drug–Gene Interactions and Adverse Drug Reactions. Pharmacogen. J. 2020, 20, 355–366.
- Joint Formulary Committee. British National Formulary (Online); BMJ and Pharmaceutical Press: London, UK, 2023; Available online: http://www.medicinescomplete.com (accessed on 27 September 2023).
- DRUG-REAX. University of Technology Sydney. Available online: https://search.lib.uts.edu.au/discovery/fulldisplay/alma991001040579705671/61UTS_INST:61UTS (accessed on 27 September 2023).
- Drug Interaction Checker. For Drugs, Food, and Alcohol. Available online: https://www.drugs.com/drug_interactions.html (accessed on 27 September 2023).
- Les Interactions Médicamenteuses. Available online: https://www.vidal.fr/medicaments/utilisation/prendre-traitement/interactions-medicamenteuses.html (accessed on 24 September 2023).
- Tatro David, S. Drug Interaction Facts: The Authority on Drug Interactions; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2014; Available online: https://books.google.it/books?id=ScqioAEACAAJ (accessed on 24 September 2023)ISBN 978-1-57439-363-7.
- Monteiro, C.; Duarte, A.P.; Alves, G. Adverse Drug Reactions in Elderly: A Five-Year Review of Spontaneous Reports to the Portuguese Pharmacovigilance System. Expert. Opin. Drug Saf. 2021, 20, 109–118.
- Dubrall, D.; Just, K.S.; Schmid, M.; Stingl, J.C.; Sachs, B. Adverse Drug Reactions in Older Adults: A Retrospective Comparative Analysis of Spontaneous Reports to the German Federal Institute for Drugs and Medical Devices. BMC Pharmacol. Toxicol. 2020, 21, 25.
- Carlquist, J.F.; Knight, S.; Horne, B.D.; Huntinghouse, J.A.; Rollo, J.S.; Muhlestein, J.B.; May, H.; Anderson, J.L. Cardiovascular Risk among Patients on Clopidogrel Anti-Platelet Therapy after Placement of Drug-Eluting Stents Is Modified by Genetic Variants in Both the CYP2C19 and ABCB1 Genes. Thromb. Haemost. 2013, 109, 744–754.
- Chan, M.Y.; Tan, K.; Tan, H.-C.; Huan, P.-T.; Li, B.; Phua, Q.-H.; Lee, H.-K.; Lee, C.-H.; Low, A.; Becker, R.C.; et al. CYP2C19 and PON1 Polymorphisms Regulating Clopidogrel Bioactivation in Chinese, Malay and Indian Subjects. Pharmacogenomics 2012, 13, 533–542.
- Harmsze, A.M.; van Werkum, J.W.; Hackeng, C.M.; Ruven, H.J.T.; Kelder, J.C.; Bouman, H.J.; Breet, N.J.; Ten Berg, J.M.; Klungel, O.H.; de Boer, A.; et al. The Influence of CYP2C19*2 and *17 on On-Treatment Platelet Reactivity and Bleeding Events in Patients Undergoing Elective Coronary Stenting. Pharmacogenet. Genom. 2012, 22, 169–175.
- Gurbel, P.A.; Shuldiner, A.R.; Bliden, K.P.; Ryan, K.; Pakyz, R.E.; Tantry, U.S. The Relation between CYP2C19 Genotype and Phenotype in Stented Patients on Maintenance Dual Antiplatelet Therapy. Am. Heart J. 2011, 161, 598–604.
- Wallentin, L.; James, S.; Storey, R.F.; Armstrong, M.; Barratt, B.J.; Horrow, J.; Husted, S.; Katus, H.; Steg, P.G.; Shah, S.H.; et al. Effect of CYP2C19 and ABCB1 Single Nucleotide Polymorphisms on Outcomes of Treatment with Ticagrelor versus Clopidogrel for Acute Coronary Syndromes: A Genetic Substudy of the PLATO Trial. Lancet 2010, 376, 1320–1328.
- Andersson, M.L.; Eliasson, E.; Lindh, J.D. A Clinically Significant Interaction between Warfarin and Simvastatin Is Unique to Carriers of the CYP2C9*3 Allele. Pharmacogenomics 2012, 13, 757–762.
- Uno, T.; Shimizu, M.; Yasui-Furukori, N.; Sugawara, K.; Tateishi, T. Different Effects of Fluvoxamine on Rabeprazole Pharmacokinetics in Relation to CYP2C19 Genotype Status. Br. J. Clin. Pharmacol. 2006, 61, 309–314.
- Sangkuhl, K.; Klein, T.E.; Altman, R.B. Clopidogrel Pathway. Pharmacogenet. Genom. 2010, 20, 463–465.
- Furuta, T.; Iwaki, T.; Umemura, K. Influences of Different Proton Pump Inhibitors on the Anti-Platelet Function of Clopidogrel in Relation to CYP2C19 Genotypes. Br. J. Clin. Pharmacol. 2010, 70, 383–392.
- Harmsze, A.M.; van Werkum, J.W.; Souverein, P.C.; Breet, N.J.; Bouman, H.J.; Hackeng, C.M.; Ruven, H.J.T.; ten Berg, J.M.; Klungel, O.H.; de Boer, A.; et al. Combined Influence of Proton-Pump Inhibitors, Calcium-Channel Blockers and CYP2C19*2 on on-Treatment Platelet Reactivity and on the Occurrence of Atherothrombotic Events after Percutaneous Coronary Intervention. J. Thromb. Haemost. 2011, 9, 1892–1901.
- Vormfelde, S.V.; Brockmöller, J.; Bauer, S.; Herchenhein, P.; Kuon, J.; Meineke, I.; Roots, I.; Kirchheiner, J. Relative Impact of Genotype and Enzyme Induction on the Metabolic Capacity of CYP2C9 in Healthy Volunteers. Clin. Pharmacol. Ther. 2009, 86, 54–61.
- Depta, J.P.; Lenzini, P.A.; Lanfear, D.E.; Wang, T.Y.; Spertus, J.A.; Bach, R.G.; Cresci, S. Clinical Outcomes Associated with Proton Pump Inhibitor Use among Clopidogrel-Treated Patients within CYP2C19 Genotype Groups Following Acute Myocardial Infarction. Pharmacogen. J. 2015, 15, 20–25.
- Laine, K.; Tybring, G.; Härtter, S.; Andersson, K.; Svensson, J.O.; Widén, J.; Bertilsson, L. Inhibition of Cytochrome P4502D6 Activity with Paroxetine Normalizes the Ultrarapid Metabolizer Phenotype as Measured by Nortriptyline Pharmacokinetics and the Debrisoquin Test. Clin. Pharmacol. Ther. 2001, 70, 327–335.
- Nicholls, G.; Youdim, K. Drug Transporters: Volume 1: Role and Importance in ADME and Drug Development; The Royal Society of Chemistry: London, UK, 2016; ISBN 978-1-78262-069-3.
- Dujic, T.; Zhou, K.; Donnelly, L.A.; Tavendale, R.; Palmer, C.N.A.; Pearson, E.R. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 2015, 64, 1786–1793.
- Ahlin, G.; Chen, L.; Lazorova, L.; Chen, Y.; Ianculescu, A.G.; Davis, R.L.; Giacomini, K.M.; Artursson, P. Genotype-Dependent Effects of Inhibitors of the Organic Cation Transporter, OCT1: Predictions of Metformin Interactions. Pharmacogen. J. 2011, 11, 400–411.
- Grün, B.; Kiessling, M.K.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Rauch, G.; Haefeli, W.E.; Czock, D. Trimethoprim-Metformin Interaction and Its Genetic Modulation by OCT2 and MATE1 Transporters. Br. J. Clin. Pharmacol. 2013, 76, 787–796.
- Wang, Z.-J.; Yin, O.Q.P.; Tomlinson, B.; Chow, M.S.S. OCT2 Polymorphisms and In-Vivo Renal Functional Consequence: Studies with Metformin and Cimetidine. Pharmacogenet. Genom. 2008, 18, 637–645.
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of Organic Cation Transporter 2 (OCT2) to Cisplatin-Induced Nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402.
- Spracklen, T.F.; Vorster, A.A.; Ramma, L.; Dalvie, S.; Ramesar, R.S. Promoter Region Variation in NFE2L2 Influences Susceptibility to Ototoxicity in Patients Exposed to High Cumulative Doses of Cisplatin. Pharmacogen. J. 2017, 17, 515–520.
- Choi, H.Y.; Bae, K.-S.; Cho, S.-H.; Ghim, J.-L.; Choe, S.; Jung, J.A.; Jin, S.-J.; Kim, H.-S.; Lim, H.-S. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 Gene Polymorphisms on the Pharmacokinetics of Simvastatin and Simvastatin Acid. Pharmacogenet. Genom. 2015, 25, 595–608.
- Luzum, J.A.; Theusch, E.; Taylor, K.D.; Wang, A.; Sadee, W.; Binkley, P.F.; Krauss, R.M.; Medina, M.W.; Kitzmiller, J.P. Individual and Combined Associations of Genetic Variants in CYP3A4, CYP3A5, and SLCO1B1 With Simvastatin and Simvastatin Acid Plasma Concentrations. J. Cardiovasc. Pharmacol. 2015, 66, 80–85.
- Tsamandouras, N.; Dickinson, G.; Guo, Y.; Hall, S.; Rostami-Hodjegan, A.; Galetin, A.; Aarons, L. Development and Application of a Mechanistic Pharmacokinetic Model for Simvastatin and Its Active Metabolite Simvastatin Acid Using an Integrated Population PBPK Approach. Pharm. Res. 2015, 32, 1864–1883.
- Nishizato, Y.; Ieiri, I.; Suzuki, H.; Kimura, M.; Kawabata, K.; Hirota, T.; Takane, H.; Irie, S.; Kusuhara, H.; Urasaki, Y.; et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) Genes: Consequences for Pravastatin Pharmacokinetics. Clin. Pharmacol. Ther. 2003, 73, 554–565.
- Pedrós, C.; Formiga, F.; Corbella, X.; Arnau, J.M. Adverse Drug Reactions Leading to Urgent Hospital Admission in an Elderly Population: Prevalence and Main Features. Eur. J. Clin. Pharmacol. 2016, 72, 219–226.
- Krishnaswami, A.; Steinman, M.A.; Goyal, P.; Zullo, A.R.; Anderson, T.S.; Birtcher, K.K.; Goodlin, S.J.; Maurer, M.S.; Alexander, K.P.; Rich, M.W.; et al. Deprescribing in Older Adults with Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 73, 2584–2595.
- Farrell, B.; Tsang, C.; Raman-Wilms, L.; Irving, H.; Conklin, J.; Pottie, K. What Are Priorities for Deprescribing for Elderly Patients? Capturing the Voice of Practitioners: A Modified Delphi Process. PLoS ONE 2015, 10, e0122246.
- Reeve, E.; Gnjidic, D.; Long, J.; Hilmer, S. A Systematic Review of the Emerging Definition of “deprescribing” with Network Analysis: Implications for Future Research and Clinical Practice. Br. J. Clin. Pharmacol. 2015, 80, 1254–1268.
- Scott, I.A.; Hilmer, S.N.; Reeve, E.; Potter, K.; Le Couteur, D.; Rigby, D.; Gnjidic, D.; Del Mar, C.B.; Roughead, E.E.; Page, A.; et al. Reducing Inappropriate Polypharmacy: The Process of Deprescribing. JAMA Intern. Med. 2015, 175, 827–834.
- Rochon, P.A.; Gurwitz, J.H. The Prescribing Cascade Revisited. Lancet 2017, 389, 1778–1780.
- Shiber, S.; Zuker-Herman, R.; Drescher, M.J.; Glezerman, M. Gender Differences in the Comprehension of Care Plans in an Emergency Department Setting. Isr. J. Health Policy Res. 2018, 7, 50.
- Kerse, N.; Buetow, S.; Mainous, A.G.; Young, G.; Coster, G.; Arroll, B. Physician-Patient Relationship and Medication Compliance: A Primary Care Investigation. Ann. Fam. Med. 2004, 2, 455–461.
- Neri, L.; Peris, K.; Longo, C.; Calvieri, S.; Frascione, P.; Parodi, A.; Eibenschuz, L.; Bottoni, U.; Pellacani, G.; the Actinic Keratosis—TReatment Adherence INitiative (AK-TRAIN) Study Group. Physician–Patient Communication and Patient-Reported Outcomes in the Actinic Keratosis Treatment Adherence Initiative (AK-TRAIN): A Multicenter, Prospective, Real-Life Study of Treatment Satisfaction, Quality of Life and Adherence to Topical Field-Directed Therapy for the Treatment of Actinic Keratosis in Italy. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 93–107.
- Hong, S.H. Potential for Physician Communication to Build Favorable Medication Beliefs among Older Adults with Hypertension: A Cross-Sectional Survey. PLoS ONE 2019, 14, e0210169.
- McInnes, G.T. Integrated Approaches to Management of Hypertension: Promoting Treatment Acceptance. Am. Heart J. 1999, 138, S252–S255.
- Shariff, Z.B.; Dahmash, D.T.; Kirby, D.J.; Missaghi, S.; Rajabi-Siahboomi, A.; Maidment, I.D. Does the Formulation of Oral Solid Dosage Forms Affect Acceptance and Adherence in Older Patients? A Mixed Methods Systematic Review. J. Am. Med. Dir. Assoc. 2020, 21, 1015–1023.e8.
- Baker, R.; Mainous Iii, A.G.; Gray, D.P.; Love, M.M. Exploration of the Relationship between Continuity, Trust in Regular Doctors and Patient Satisfaction with Consultations with Family Doctors. Scand. J. Prim. Health Care 2003, 21, 27–32.
- Ridd, M.; Shaw, A.; Lewis, G.; Salisbury, C. The Patient–Doctor Relationship: A Synthesis of the Qualitative Literature on Patients’ Perspectives. Br. J. Gen. Pract. 2009, 59, e116–e133.
- Thom, D.H.; Kravitz, R.L.; Bell, R.A.; Krupat, E.; Azari, R. Patient Trust in the Physician: Relationship to Patient Requests. Fam. Pract. 2002, 19, 476–483.
This entry is offline, you can click here to edit this entry!