Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Thiols are one of the most convenient synthons in the synthesis of organosulfur compounds. The typical methods to prepare monoterpene thiols include the electrophilic addition of H2S or dithiols to the double bond of monoterpenes; nucleophilic substitution of halides; tosylates/mesylates obtained from corresponding monoterpene alcohols; thia-Michael addition of S-nucleophiles to α,β-unsaturated ketones; nucleophilic epoxide ring opening; nucleophilic substitution of the activated methylene protons; and reduction of sulfochlorides, dithiolanes, thiiranes, and sultones.
- monoterpenoids
- thiols
- asymmetric synthesis
- disulfides
- thiosulfonates
1. Synthesis from Alkenes
The synthesis of terpene thiols from limonene, α-pinene, α-, γ-terpinenes, terpinolene, and 3-carene via a reaction of them with H2S in the presence of Lewis acids such as AlCl3 or AlBr3 is described in [1]. The addition of H2S usually occurs without selectivity and is accompanied by numerous side reactions, including the rearrangement of the terpene skeleton, especially in cases with bicyclic systems. The addition of H2S to limonene 1 catalyzed by AlCl3 proceeds with no regioselectivity and gives thiols 2–5 in low yields, with the intramolecular cyclization of thiols 4 and 5 at the double bond affording sulfides 6 and 7 as the main products (Scheme 1) [2][3][4].

Scheme 1. The addition of H2S to limonene 1 catalyzed by AlCl3.
The interaction of α-pinene 8 with H2S under the same conditions leads to products 2–7, as well as cyclic sulfide 9 [1].
Electrophilic thiylation of α-pinene 8 with H2S in the presence of AlBr3 (A) is followed by the pinene–menthane rearrangement, providing carbocation 10, which, when reacting with H2S, gives thiol 4. The softer Lewis acid EtAlCl2 (B) stereoselectively catalyzes the anti-addition of H2S via the formation of intermediate 11 and leads to trans-pinane-2-thiol 12 (Scheme 2) [5]. With a strong Lewis acid (BF3·Et2O) used as a catalyst, the Wagner–Meerwein rearrangement occurs to yield isobornanethiol 13 [3][5].
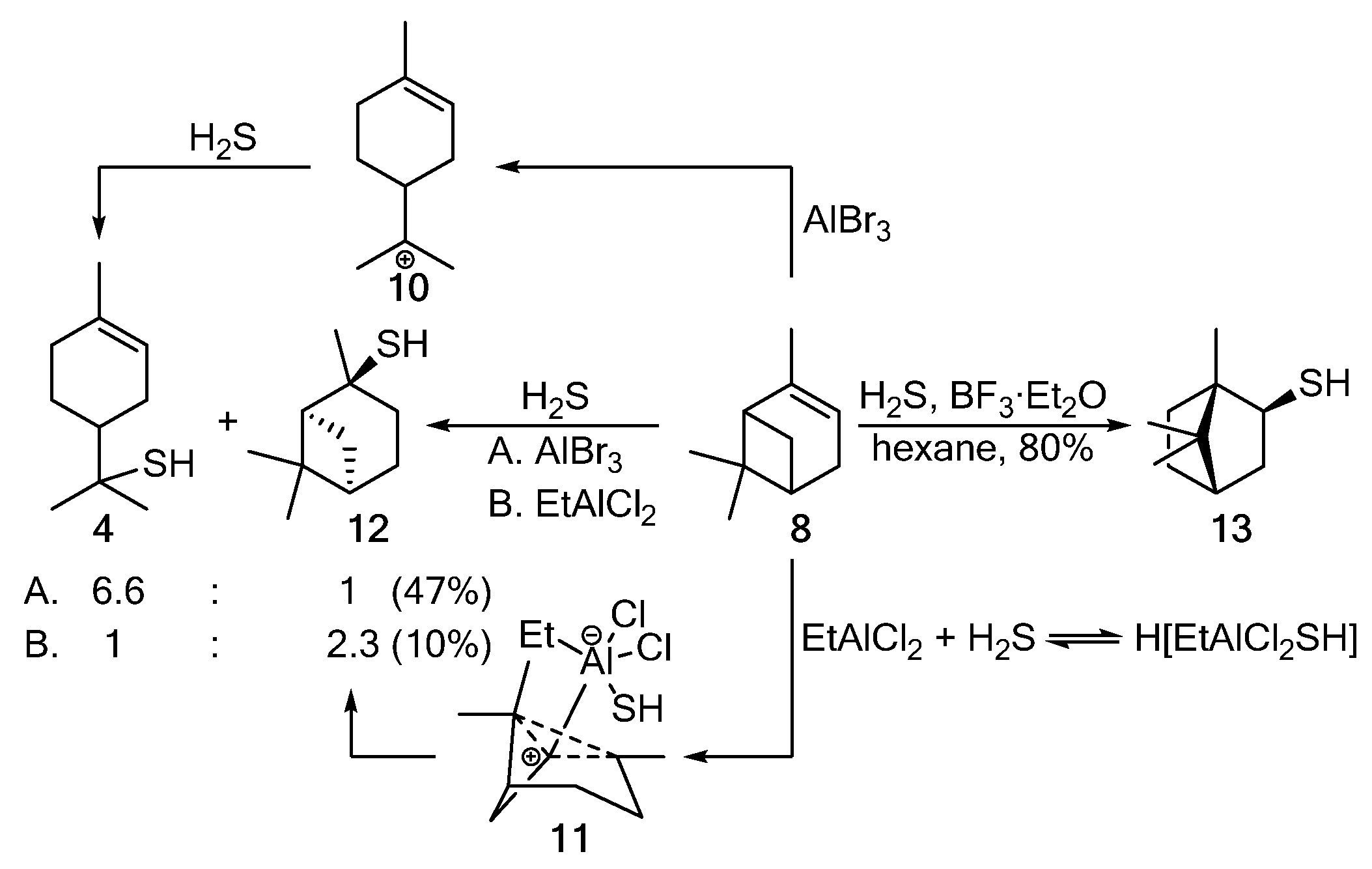
Scheme 2. The addition of H2S to α-pinene 8.
The addition of hydrogen sulfide to 3-carene 14 in the presence of AlCl3 proceeds nonselectively to give the products in low yields. The detected products included a mixture of cis- and trans-thiols 15; episulfides 16, 6, and 7; and para-menthane thiols 17, 18, 2, and 3 (Scheme 3) [1].
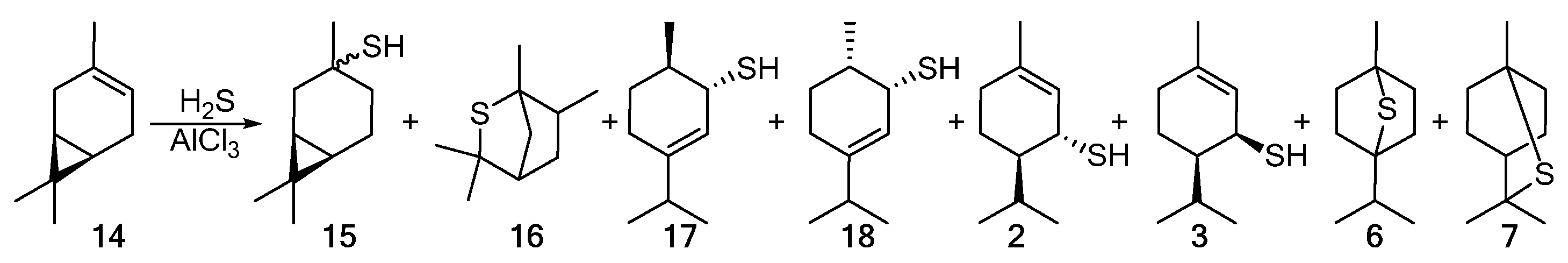
Scheme 3. The addition of H2S to 3-carene 14 catalyzed by AlCl3.
Reactions of racemic camphene 19 with thioacetic acid under various conditions were investigated in [6] (Scheme 4). It was established that, under catalyst-free conditions and with a long reaction time (12 h), the anti-Markovnikov product 20 was predominantly formed. The use of p-toluenesulfonic acid as a catalyst also leads to thioester 20, but in a 15% yield. Catalysis with trifluoromethanesulfonic acid (TfOH) and InCl3 at different temperatures gives different ratios of products. The optimal yield of thioacetate 21 (75%), a product of the Wagner–Meerwein rearrangement, was achieved using a catalyst TfOH at 40 °C for 20 min. The yield of a by-product, thioacetate 20, from this procedure does not exceed 25%. The best method to obtain Markovnikov product 22 (82%) with a preserving camphane structure was catalysis via In(OTf)3 at ≤0 °C. The deacylation of thioacetate 22 with LiAlH4 leads to racemic camphane thiol 23 at an 86% yield.
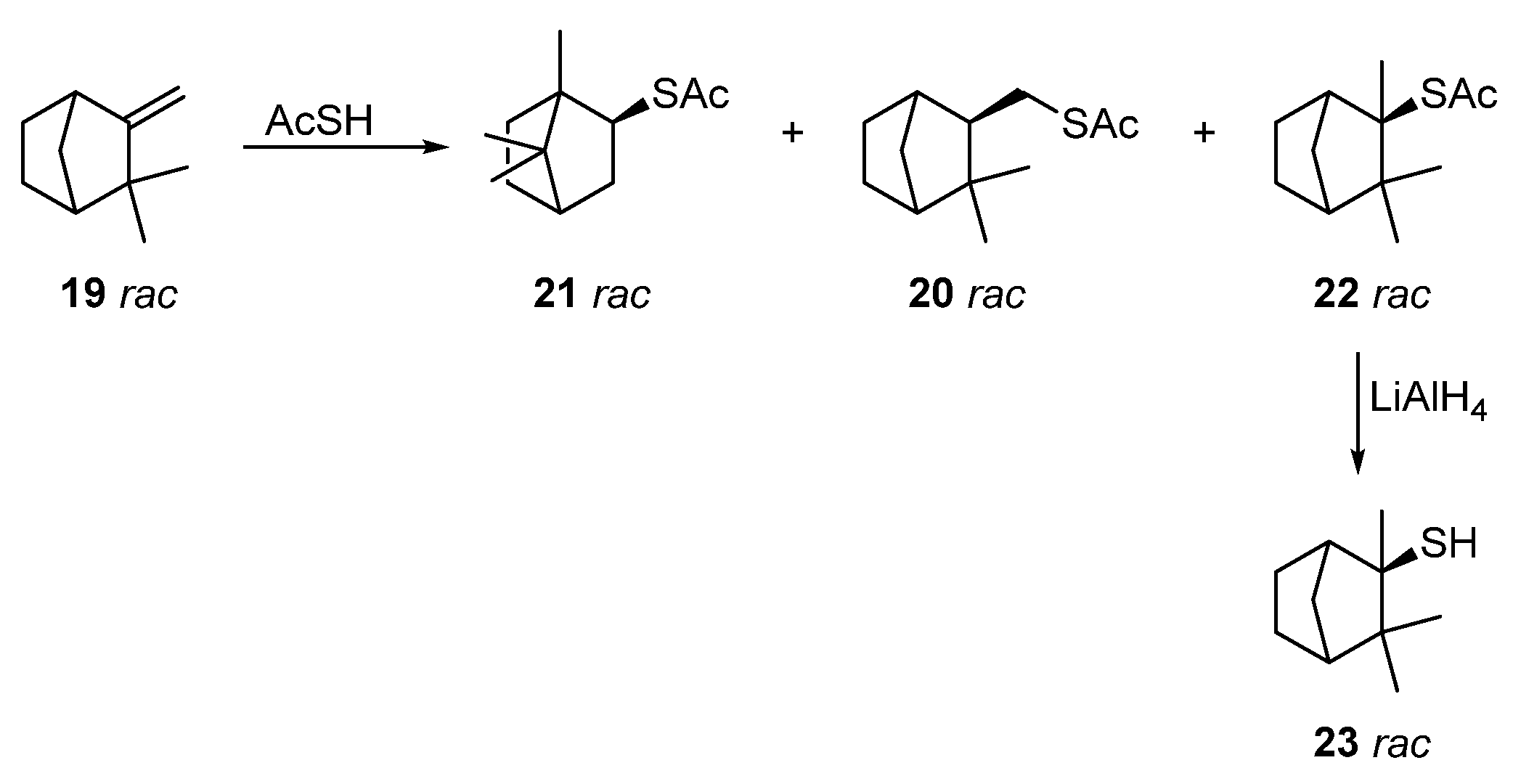
Scheme 4. Synthesis of camphane thiol 23.
Photochemical addition of thioacetic acid to (−)-sabinene 24 gives a mixture of anti-Markovnikov bicyclic thioacetate 25 and unsaturated thioacetate 26 in an overall yield of 24% and a 3:1 ratio, respectively [7]. The unexpected formation of thioacetate 26 results from cyclopropane ring cleavage. The mixture of thioacetates 25 and 26 was treated with LiAlH4 to produce thiols 27 and 28 in an overall yield of 95% (Scheme 5). The obtained thiols were isolated by preparative capillary GC.

Scheme 5. Synthesis of thiols from sabinene 24.
2. Ene Reaction of Monoterpenes with N-sulfinylbenzenesulfonamide
An efficient method for the synthesis of monoterpene allyl thiols using N-sulfinylbenzenesulfonamide 29 as an enophile in ene reaction was proposed in the paper [8] (Scheme 6). The interaction of terpenes (α- and β-pinenes 8 and 30; 2- and 3-carenes 31 and 14; and α-thujene 32) with N-sulfinylbenzenesulfonamide 29 proceeds at a double bond with the formation of adducts 33–37 with a migration of the double bond to an α-position. It should be noted that these reactions occur stereo- and regioselectively. The adducts 33–37, when reduced with LiAlH4, provide the corresponding allyl thiols, 38–42.

Scheme 6. Synthesis of allylic terpene thiols 38–42.
3. Synthesis from α,β-Unsaturated Carbonyl Compounds
Thiols are good nucleophiles for thia-Michael addition to α,β-unsaturated carbonyl compounds [9]. However, harsh reaction conditions are required to convert the newly formed sulfide group into a synthetically more versatile SH group. Thioacids (RCOSH) are more attractive as nucleophiles for the Michael addition reaction, since the resulting thioesters can be easily transformed into corresponding thiols under mild conditions [10][11][12].
Myrtenal-based hydroxythiol 43 was synthesized by two methods with a high yield and stereoselectivity [10]. The treatment of (−)-myrtenal 44 with benzylthiol and 10% aqueous NaOH in THF at room temperature for 18 h led to sulfide 45 (yield 92%, de 96%). Compound 45 was reduced to the corresponding alcohol 46 (yield 96%) with LiAlH4 in Et2O, which was then hydrogenolyzed to hydroxythiol 43 under Birch reduction conditions (Scheme 7). The hydrogenolysis did not provide satisfactory results because small differences in reaction conditions altered the reaction course dramatically, sometimes producing a complex mixture of unidentified compounds. The same reaction conditions become reproducible in switching to thioacetic acid as a nucleophilic reagent, which demonstrated a high selectivity when added to (−)-myrtenal 44 to give thioacetate 47 (1,4-addition) in yield of 98% and de > 99%. Thioester 47 was reduced by LiAlH4 to obtain hydroxythiol 43 in a 95% yield. This one-pot method allowed us to simultaneously convert thioether and aldehyde group to the corresponding thiol and primary alcohol (Scheme 7).
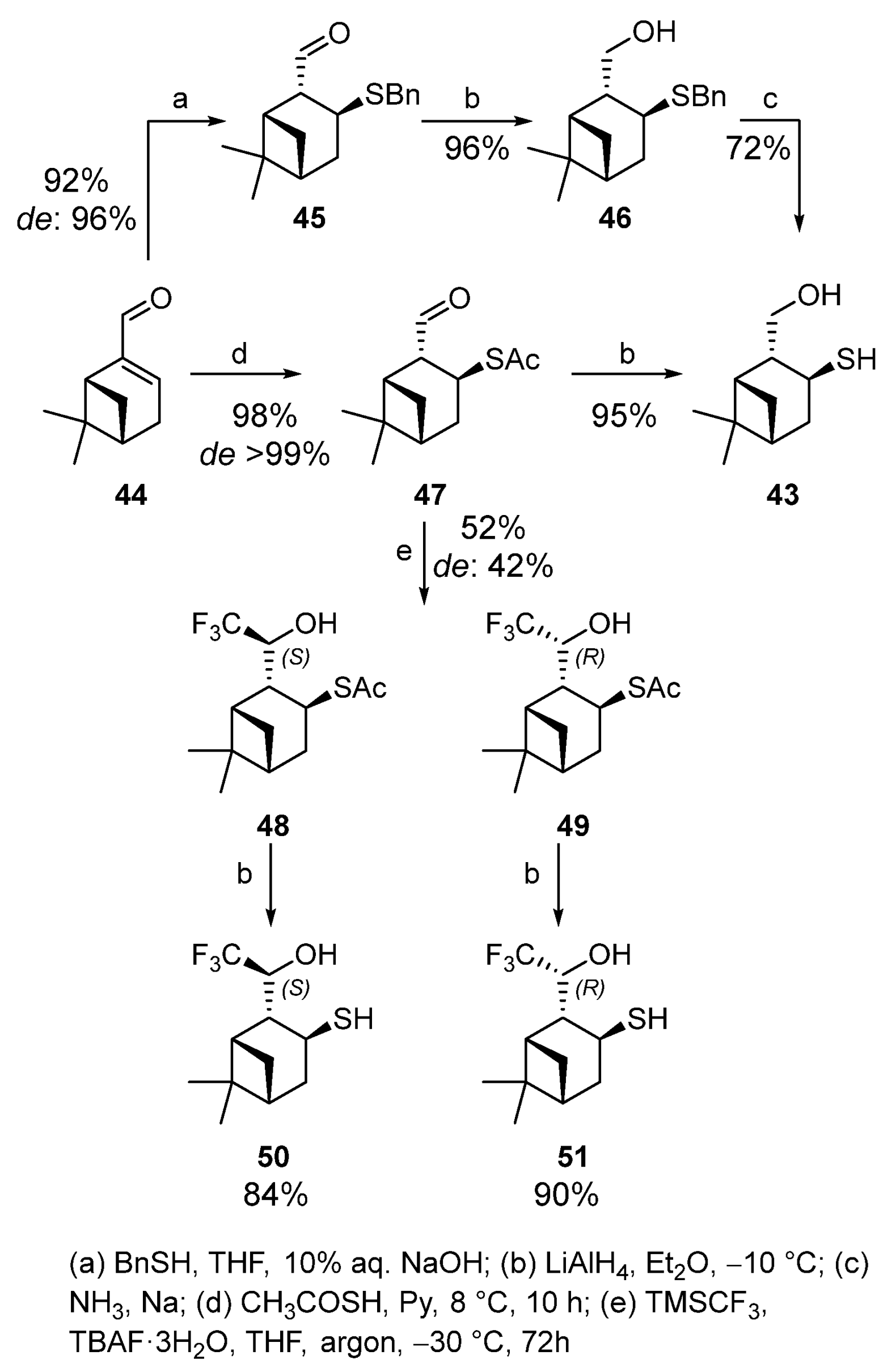
Scheme 7. Synthesis of pinane hydroxythiols based on myrtenal 44.
Trifluoromethylation of 2-formylisopinocampheyl-3-thioacetate 47 by Ruppert–Prakash reagent in the presence of tetra-n-butylammonium fluoride (TBAF) was carried out at −30 °C for 3 days. Diastereomers 48 and 49 are formed in a 52% total yield and de 42% with the predominance of thioacetate 48. Deacylation of thioacetates 48 and 49 with LiAlH4 in dry Et2O under an argon atmosphere gives the corresponding thiols 50 and 51 with 84 and 90% yields, respectively (Scheme 7) [13].
Thioacetate 52 was obtained from (1S)-(−)-verbenone 53 by using a procedure similar to the synthesis of 2-formylisopinocampheyl-3-thioacetate 47. The reaction produces one of two theoretically possible diastereomers with the R-configuration of C-2 with a 71% yield (Scheme 8). Thioacetate 52 does not react with the Rupert–Prakash reagent under the above conditions, possibly because of the bulky TBAF use.
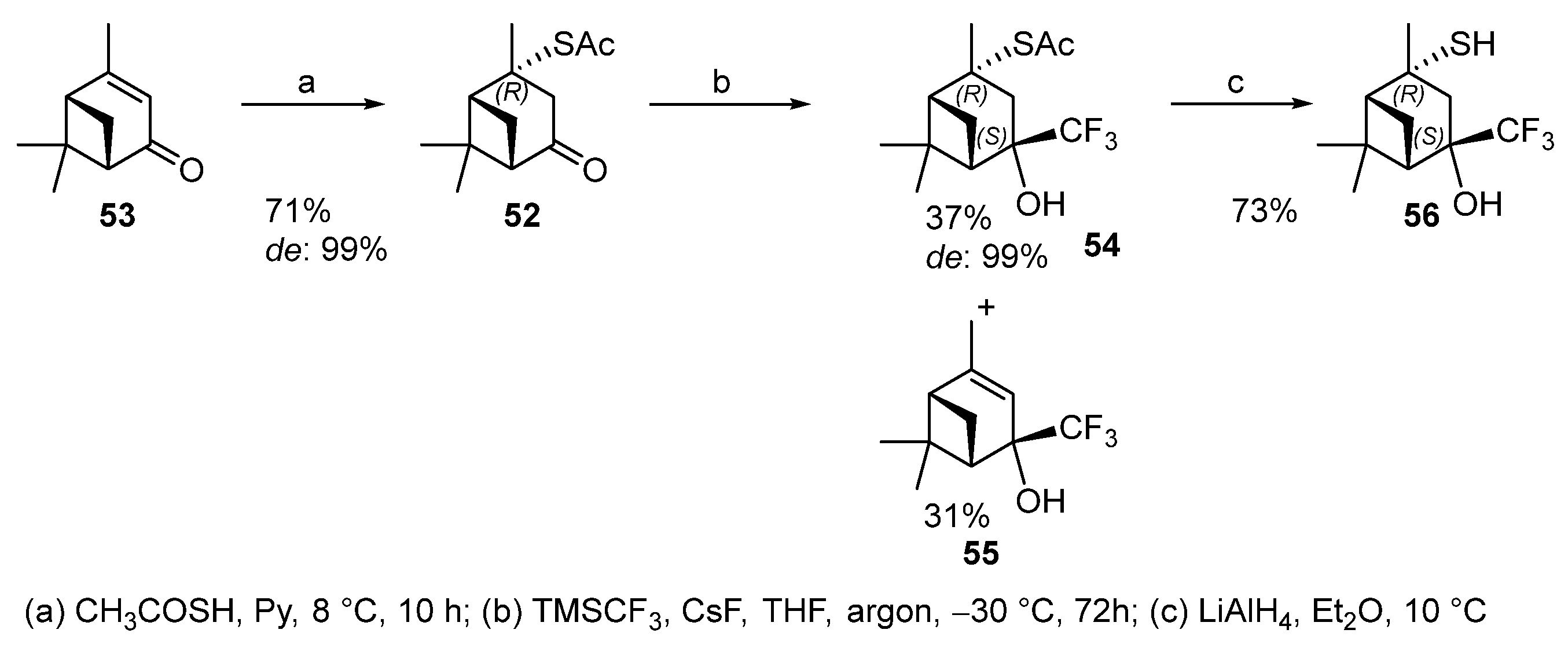
Scheme 8. Synthesis of pinane hydroxythiols based on verbenone 53.
The addition of fluorine-containing initiator CsF made it possible to obtain the only (4S)-diastereomer 54 in a 37% yield together with trifluoromethyl alcohol 55 (31%) that is a by-product of desulfurization (Scheme 8). Deacylation of thioacetate 54 gave hydroxythiol 56 in 73% yield [13].
The synthesis of isomeric hydroxythiols 57–59 was carried out on the basis of β-pinene 30 (Scheme 9) [14]. Trans-pinocarveol 60 was synthesized via the oxidation of β-pinene 30 with the SeO2/TBHP system, and its further oxidation with MnO2 led to pinocarvone 61. An inseparable mixture of two isomeric ketothioacetates (2S)-62 and (2R)-63 in a 2:1 ratio in 95% yield is formed during the thia-Michael reaction of pinocarvone 61 with AcSH in the presence of catalytic amount of pyridine at −5 °C. The reduction of thioacetates with LiAlH4 leads to three isomeric hydroxythiols, 57–59.

Scheme 9. Synthesis of pinane hydroxythiols based on β-pinene 30.
The synthesis of pinane ketothiols 64 and 65 was implemented from α,β-unsaturated pinane ketones 61 and 66 [15]. To obtain thioacetate 62 from enone 61, the synthetical protocol proposed in [10] was used. However, the diastereoselectivity of this reaction under the described conditions did not exceed 33%, as mentioned in [14]. The de value of thioacetate 62 can be increased from 33 up to 92% if the reaction between pinocarvone 61 and AcSH is carried out in THF in a temperature range from −60 to −65 °C, with pyridine as a co-solvent. The same conditions are applicable for the addition of BzSH to ketone 61, with thioacetate 67 being formed in this case with a comparable de of 93% (Scheme 10). Reducing thioacetate 62 via NH2NH2·H2O affords thiol 64 within 4-5 h in up to a 90% yield, while deacylation of thiobenzoate 67 by the same reagent gives the thiol in only a 38-50% yield due to incomplete conversion. Thus, at comparable maximum de values of thioesters 62 and 67, the preparation of thiol 64 from compound 62 is more optimal, taking into account the higher total yield of thiol and the diacylation time.

Scheme 10. Synthesis of β-ketothiol from pinocarvone 61.
A multistep synthesis of 2-norpinanone 66 from (−)-β-pinene 30 was provided in [16] (Scheme 11). This compound was obtained via nopinone 69 and then ketoenol 68 formation. Ketoenol 68 was produced in a 96% yield from ketone 69 by its reaction with isoamyl formate and t-BuOK in THF at 0 °C for 6 h [15]. The following dihydroxylation of ketoalcohol 68 by formaldehyde in sodium carbonate solution afforded 2-norpinanone 66 [15]. An addition of thioacetic acid to 2-norpinanone 66 was, for the first time, implemented according to the procedure [10] and then by using pyridine as a catalyst [9] in THF at room temperature [15]. The main product of this reaction was the isomer (3R)-70 (de 98%) (Scheme 11). Its deacylation by hydrazine hydrate (NH2NH2·H2O) led to 2-ketothiol 65 and disulfide 71 in a 3:1 ratio, respectively. Because of the mild reducing properties of NH2NH2·H2O and its inability to donate protons, the diacylation proceeds chemoselectively with the preservation of the carbonyl group [17], a behavior that is not typical for LiAlH4 when used [14].

Scheme 11. Synthesis of β-ketothiol based on 2-norpinanone 66.
Pulegone 73 was used to synthesize para-menthane-derived β-hydroxythiol 72 (Scheme 12) [18][19][20][21]. The 1,4-addition of sodium benzyl thiolate to pulegone led to a diastereomeric mixture of ketosulfides 74 in a 4:1 ratio. Then, the mixture 74 was reduced under Birch conditions by Na in liquid NH3 to give a mixture of hydroxythiols 72. Condensation of 72 with benzaldehyde and subsequent crystallization from acetone afforded diastereomerically pure oxathiane 75 in a 50% yield. When oxidized by AgNO3 in the presence of NCS, oxathiane 75 is transformed into sultines 76, the reduction of which with LiAlH4 gives pure β-hydroxythiol 72.
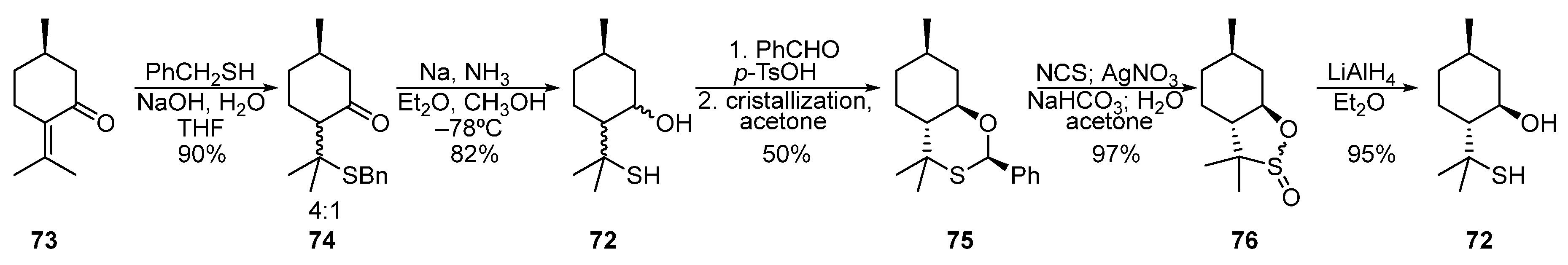
Scheme 12. Synthesis of β-hydroxythiol based on pulegone 73.
Isomeric α,β-hydroxythiols 77 and 78 were obtained from natural 3-carene 14 (Scheme 13) [22]. 3-Carene, when oxidized by m-CPBA, selectively forms trans-epoxide 79, which is isomerized in the presence of diethylaluminum 2,2,6-tetramethylpiperidide (DATMP) to enol 80 [23]. The oxidation of alcohol 80 to enone 81 is successfully implemented by the bis(acetoxy)iodobenzene (BAIB)–2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) system. Enone 81, being an unstable compound, cannot be isolated in its pure form. The two-step thia-Michael addition of AcSH to α,β-unsaturated ketone 81 proceeds in one pot in pyridine. As a result, only one of the two theoretically possible diastereomers, thioacetate 82, is formed. The subsequent reduction of ketothioacetate 82 by LiAlH4 leads to two diastereomeric β-hydroxythiols, 77 and 78, in a 1:2 ratio, respectively [22].
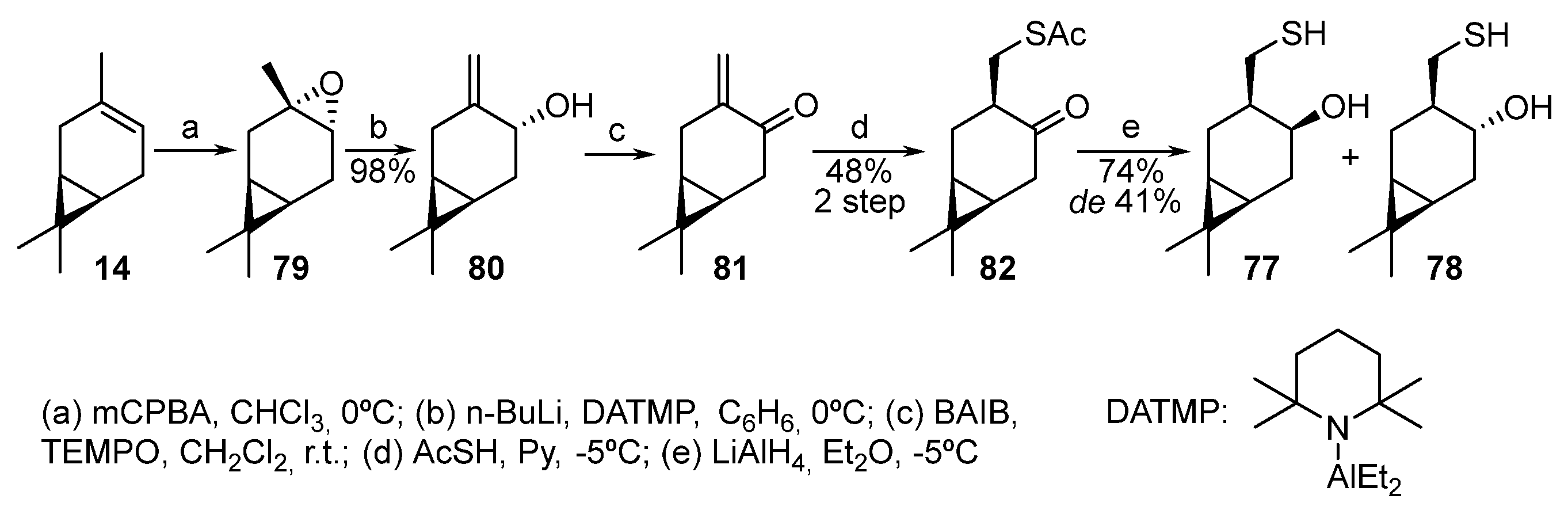
Scheme 13. Synthesis of monoterpene hydroxythiols based on 3-carene 14.
4. Synthesis from Alcohol via Tosylates, Halides, Isothiouronium Salts
The works [24][25][26][27] cover the methods for the selective preparation of neomenthanethiol 83 using thioacetic acid (AcSH) (Scheme 14). Starting menthol 84 reacts with p-TsCl in pyridine to form tosylate 85, which, when heated with AcSK, gives thioacetate 86 in a 77% yield. Substitution of the OTs (p-toluenesulfonate, tosylate) by the AcS-group occurs with an inversion of the chiral center via the SN2 mechanism. The reduction of 86 by LiAlH4 provides diastereomerically pure thiol 83 in a 26–40% yield (Scheme 14).
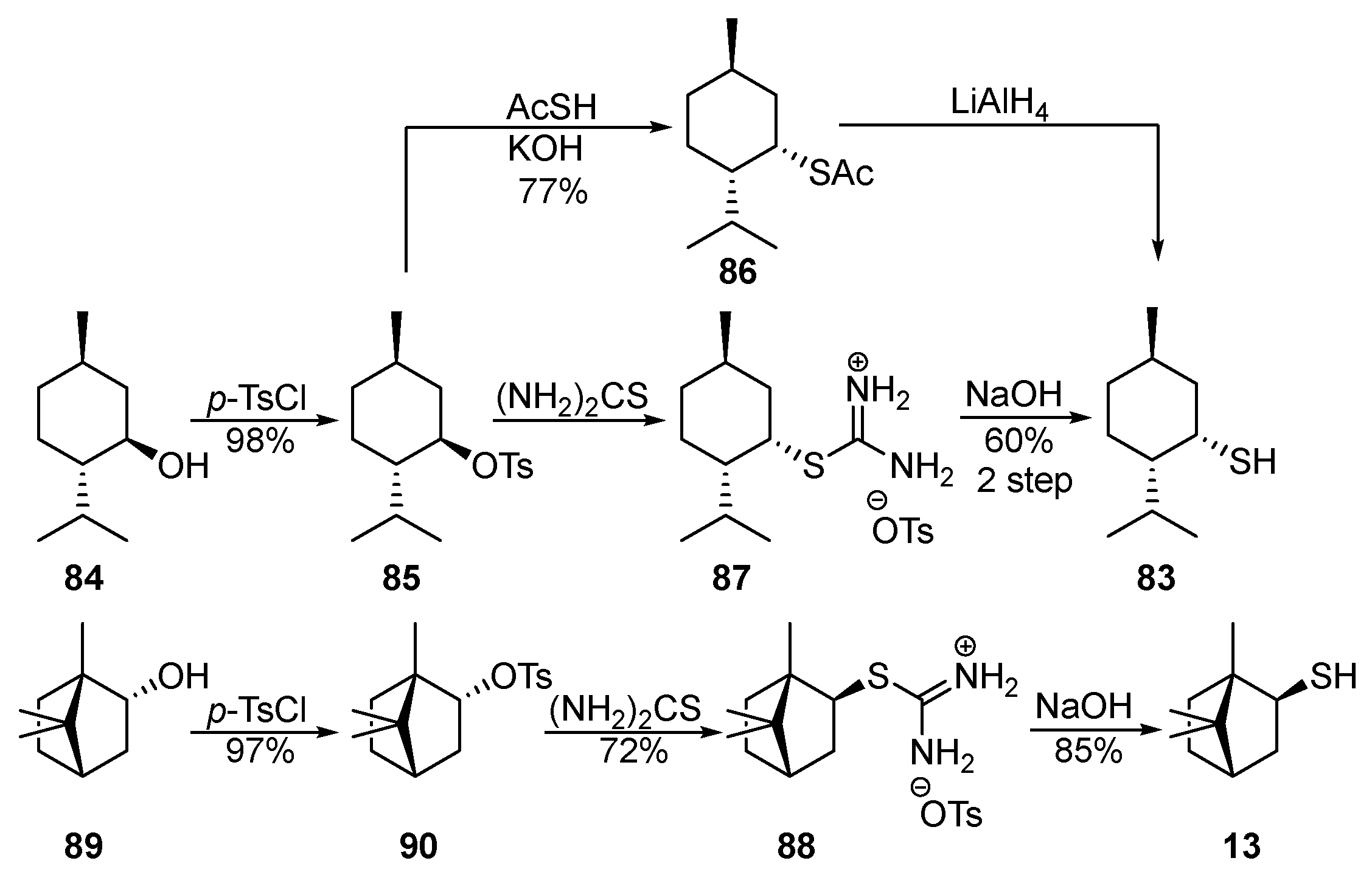
Scheme 14. Synthesis of neomenthanethiol 83 and isobornanethiol 13.
Neomenthanethiol 83 [27][28] and isobornanethiol 13 [27][29][30][31] were also synthesized in good yields via isothiouronium salts 87 and 88, proceeding from alcohols 84 and 89 (Scheme 14).
In addition to neomenthanethiol 83 and isobornanethiol 13, the authors of [27] prepared 4-caranethiol 91 and cis-myrtanethiol 92 using the same method.
(−)-(3R)-Pinanthiol 93 was proposed to be obtained via the Mitsunobu-Rollin procedure from (+)-isopinocampheol 94 [32][33][34] (Scheme 15). The reaction of the alcohol 94 with zinc N,N-dimethyldithiocarbamate in the presence of triphenylphosphine and diethylazodicarboxylate (DEAD) is accompanied by an inversion of C-3 configuration and leads to dithiocarbamate 95 in a 66% yield. Dithiocarbamates baced on menthol 84 and borneol 89 were also obtained by the same original procedure [34][35]. The reduction of dithiocarbamate 95 by LiAlH4 gives thiol 93 in a 92% yield. The approach to obtain thiol 93 through the corresponding mesylate 96 and thioacetate 97 was described in [36].
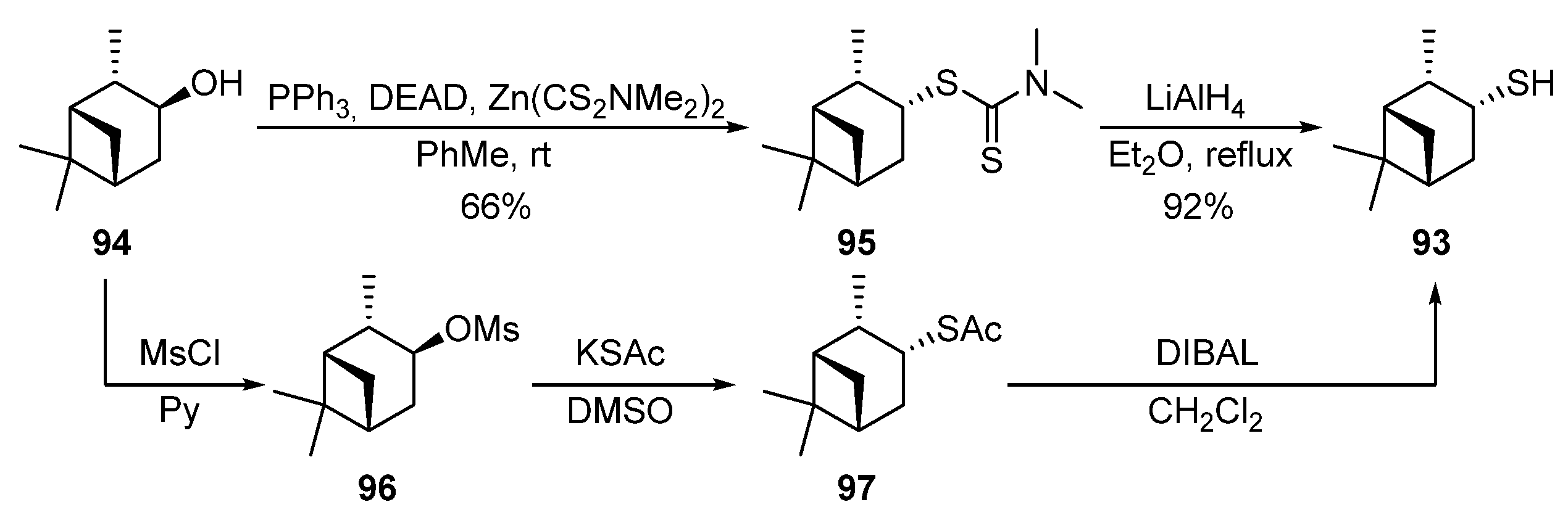
Scheme 15. Synthesis of (1S,2S,3R,5R)-3-pinanethiol 93.
Geraniol 98 reacts with thioacetic acid under Mitsunobu-type conditions [37] to form thioacetate 99 in a good yield, which, when treated with LiAlH4, is converted into the corresponding thiol 100 in a 61% yield (Scheme 16) [38].
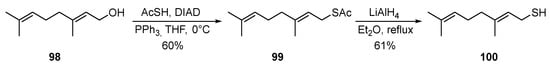
Scheme 16. Synthesis of thiogeraniol 100.
The ability of nerol 101 to be converted into bromide 102 under the action of PBr3, and then into thiol 103 by using NaSH via two successive nucleophilic substitutions with yields of 86 and 66%, respectively, was described in [39] (Scheme 17).
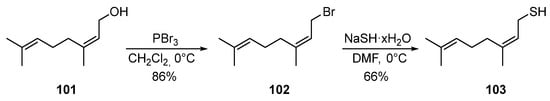
Scheme 17. Synthesis of thionerol 103.
Diastereomerically pure hydroxythiol 57 can also be obtained via two alternative routes [14]. The first one involves the bromination of β-pinene 30 by NBS (N-bromosuccinimide) to form myrtenyl bromide 104, which undergoes hydroboration–oxidation and is selectively transformed to bromoalcohol 105. The nucleophilic replacement of bromide by thioacetate AcS− leads to compound 106, which can also be synthesized starting from α-pinene 8 (Scheme 18). The second route is associated with the oxidation of α-pinene 8 to myrtenal, followed by its reduction to myrtenol 107, which is converted into diol 108 by the same hydroboration–oxidation procedure. The further reaction of tosyl chloride with diol 108 leads to both monotosylate 109 (76%) and ditosylate 110 (10%). The nucleophilic substitution of the para-toluenesulfonate group in 109 by AcS− also results in thioacetate 106. When reduced, thioacetate 106 affords hydroxythiol 57 (Scheme 18) [14].

Scheme 18. Synthesis of 10-hydroxyisopinocampheylthiol 57 from α- and β-pinene.
5. Nucleophilic Substitution of the Activated Methylene Proton
The synthesis of bornane α-hydroxythiol 111 was described in [40][41] (Scheme 19). The nucleophilic substitution of a proton of the activated methylene group in camphor 112 by benzyl p-toluenesulfonate promoted by LDA leads to the formation of ketosulfide 113, which, being reduced by NaBH4 in methanol or dibutylaluminum hydride (DIBAL) in THF, gives hydroxysulfide 114, which is capable of being transformed into hydroxythiol 111 by the Birch reduction.
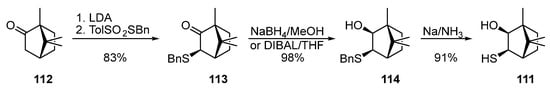
Scheme 19. Synthesis of bornane α-hydroxythiol 111 from camphor 112.
6. Epoxide and Thiiran Ring Opening
The nucleophilic ring opening of epoxide 79 with AcSH catalyzed by tetramethylammonium fluoride (TMAF) yields hydroxythioacetate 115, which is readily deacylated by LiAlH4 to form the corresponding α-hydroxythiol 116 (Scheme 20).

Scheme 20. Synthesis of monoterpene hydroxythiols 116 and 120 based on 3-carene 14.
Cis-epoxide 117 was obtained according to the known method [42] through bromohydrin 118 in 70% total yield. The interaction of epoxide 117 with AcSH in the presence of TMAF leads to thioacetate 119, the deacylation of which gives α-hydroxythiol 120 (Scheme 20) [22].
The nucleophilic sulfenylation of carane thiiranes, cis-121 and trans-122, by mono- (MeSH, EtSH, n-BuSH, PhSH) and bifunctional (HSCH2CH2OH) thiols, promoting with sodium ethoxide and thiolates, affords mercaptosulfides 123–128 with only moderate yields. By-product disulfides 129 and 130 are additionally formed during the reaction of thiiranes 121 and 122 with 2-mercaptoethanol (Scheme 21) [43].

Scheme 21. Sulfenylation of carane thiiranes 121 and 122.
7. Reduction of Thiiranes, Thiolanes, Sulfonyl Chlorides, and Sultones
Monoterpene thiols can be obtained via the reduction of thiiranes. A method for the directed synthesis of racemic thiol 4 from thiirane 131 through oxirane 132 and isothiouronium salt 133 was described in [4]. The sequential reflux of epoxide 132 with thiourea and Na2CO3 leads to the corresponding thiirane 131, the reduction of which by LiAlH4 gives thiol 4 in a moderate yield. A similar protocol for obtaining racemic thiol 5 was reported in [44]; however, thiiran 134 in this study was synthesized from oxirane 135 using the N,N-dimethylthioformamide (DMTF)–TFA system as a reagent (Scheme 22).

Scheme 22. Scheme for the synthesis of racemic 1-p-menthene-8-thiol 4 and 1-p-menthene-4-thiol 5.
Trans-limonene-1,2-epoxide 137 and cis-1,2-limonene-1,2-epoxide 138 were transformed by the DMTF-TFA system into cis-139 and trans-1,2-epithio-p-ment-8-ene 140, respectively (Scheme 23) [45]. The yield of thiirane 140 is lower than that of thiirane 139, since the reaction is accompanied by the formation of the by-product diol 141, which is yielded during the acid hydrolysis of epoxide 138. The reductive cleavage of the thiirane ring of 139 proceeds readily to give thiols 142 and 143, of which only thiol 142 was isolated in its pure form. Thiirane 140 was proposed to reduce to thiol 144 at only a 37% yield.

Scheme 23. Synthesis and reduction of para-menthane thiiranes 139 and 140.
Thioketals can also be used as the starting compounds for the synthesis of monoterpene thiols. Thus, the reductive cleavage of menthone dithiolane 145 using n-BuLi leads to the diastereomeric mixture of menthanethiol 146 and neomenthanethiol 83 (Scheme 24) (A) [46], (B) [47].
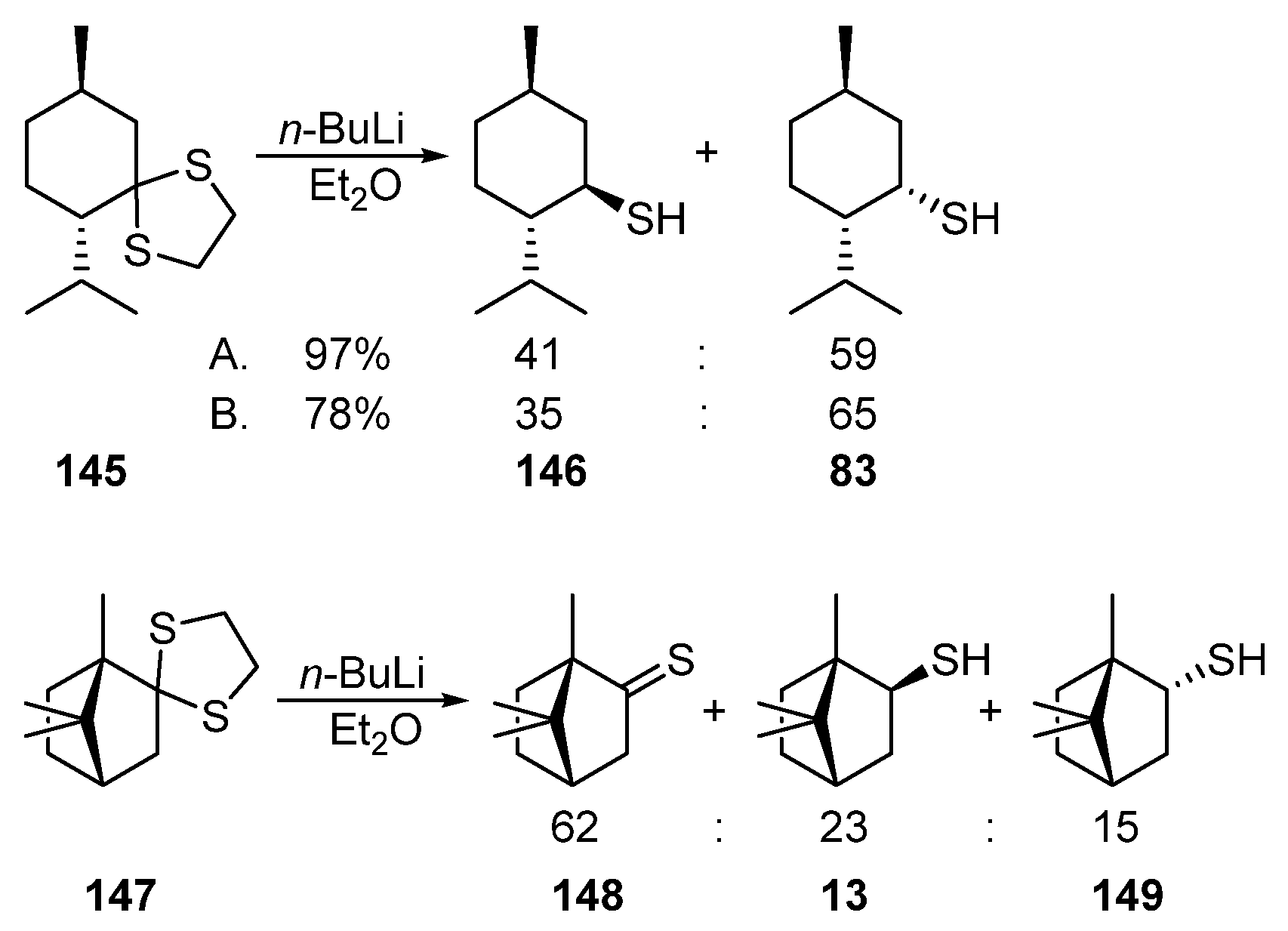
Scheme 24. Reductive cleavage of menthone dithiolane 145 and camphor dithiolane 147.
The reductive cleavage of camphor dithiolane 147 induced by n-BuLi produces thiocamphor 148 (62%) as the major product; the mixture of exo-13 and endo-149 thiols accounts for only 38% (Scheme 24) [48].
Some methods to obtain bornane β-hydroxythiols 150 and 151 by reducing camphor-10-sulfonyl chloride 152 are described in [49][50][51]. As a result of this transformation, two diastereomeric hydroxythiols, 150 and 151, are formed (Scheme 25). Camphor-10-sulfonyl chloride 152 can also be selectively converted into ketothiol 153 by using PPh3 as a reducing agent [52][53].
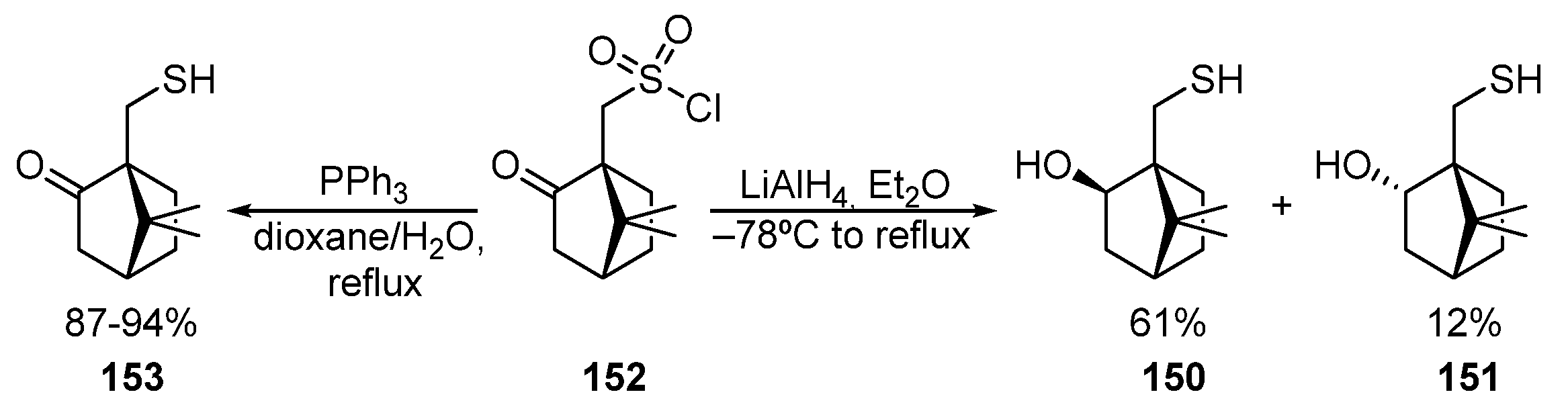
Scheme 25. Synthesis of 10-thioisoborneol 150, 10-thioborneol 151, and 10-thiocamphor 153.
The authors of [54][55] carried out the reduction of bornane sultones 154 and 155 by LiAlH4 in THF to form the corresponding mixture of hydroxythiols 156 and 150, sultines 157 and 158, borneol 89, and isoborneol 159 (Scheme 26).
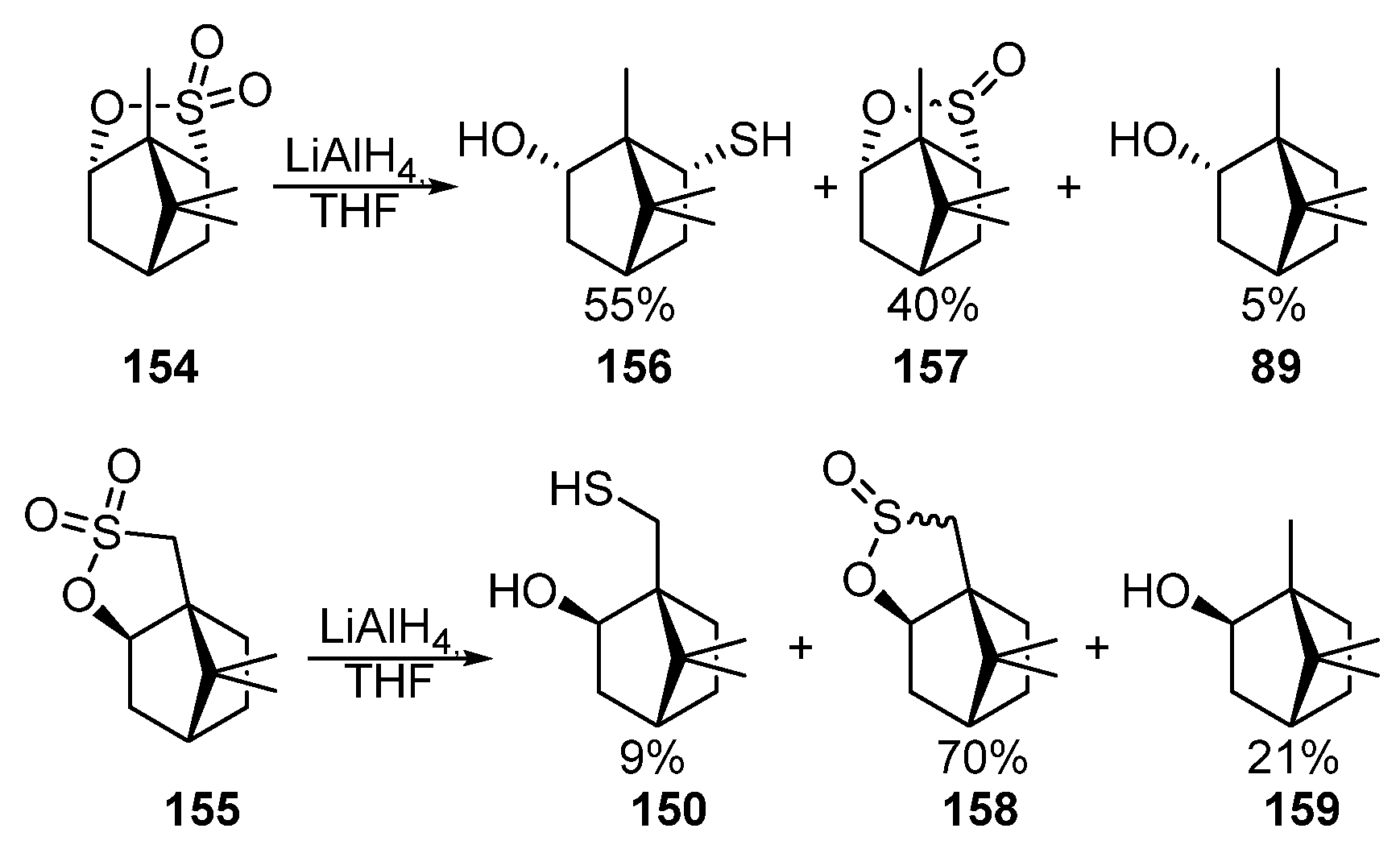
Scheme 26. Reduction of monoterpene sultones 154 and 155.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115884
References
- Janes, J.F.; Marr, I.M.; Unwin, N.; Banthorpe, D.V.; Yusuf, A. Reaction of Monoterpenoids with Hydrogen Sulphide to Form Thiols Andepi-Sulphides of Potential Organoleptic Significance. Flavour Fragr. J. 1993, 8, 289–294.
- Tolstikov, G.A.; Kanzafarov, F.Y.; Sangalov, Y.A.; Dzhemilev, U.M. Alkylaluminum Chlorides as Catalysts of the Addition of Hydrogen Sulfide to Olefins. Neftekhimiya 1979, 19, 425–429.
- Tolstikov, G.A.; Kanzafarov, F.Y.; Dzhemilev, U.M.; Kantyukova, R.G.; Zelenova, L.M. Electrophilic Thiylation of Bicyclic Monoterpenes by Hydrogen Sulfide and 1-Butanethiol. Zhurnal Organicheskoi Khimii 1983, 19, 2075–2081.
- Demole, E.; Enggist, P.; Ohloff, G. 1-p-Menthene-8-thiol: A powerful flavor impact constituent of grapefruit juice (Citrus parodisi MACFAYDEN). Helv. Chim. Acta 1982, 65, 1785–1794.
- Goeke, A. Sulfur-Containing Odorants in Fragrance Chemistry. Sulfur Rep. 2002, 23, 243–278.
- Weïwer, M.; Chaminade, X.; Bayón, J.C.; Duñach, E. Indium Triflate-Catalysed Addition of Thio Compounds to Camphene: A Novel Route to 2,3,3-Trimethyl-2-thiobicyclo[2.2.1]Heptane Derivatives. Eur. J. Org. Chem. 2007, 2007, 2464–2469.
- Candela, K.; Fellous, R.; Joulain, D.; Faure, R. An Unusual Rearrangement of the Thujane Skeleton under Free-Radical Conditions: Synthesis of Thioacetates and Thiols Derivatives of (-)-Sabinene. Sulfur Lett. 2002, 25, 145–149.
- Gadras, A.; Dunogues, J.; Calas, R.; Deleris, G. Regiospecific Two-Step Synthesis of Optically Active Allylic Terpenyl Thiols. J. Org. Chem. 1984, 49, 442–444.
- Wadhwa, P.; Kharbanda, A.; Sharma, A. Thia-Michael Addition: An Emerging Strategy in Organic Synthesis. Asian J. Org. Chem. 2018, 7, 634–661.
- Martínez-Ramos, F.; Vargas-Díaz, M.E.; Chacón-García, L.; Tamariz, J.; Joseph-Nathan, P.; Zepeda, L.G. Highly Diastereoselective Nucleophilic Additions Using a Novel Myrtenal-Derived Oxathiane as a Chiral Auxiliary. Tetrahedron Asymmetry 2001, 12, 3095–3103.
- Kambe, N. Product Class 5: Alkanethiols. In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany, 2015; Volume 39, ISBN 978-3-13-118921-9.
- Shcherbakova, I.; Pozharskii, A.F. Alkyl Chalcogenides: Sulfur-Based Functional Groups. In Comprehensive Organic Functional Group Transformations II; Elsevier: Amsterdam, The Netherlands, 2005; pp. 89–235. ISBN 978-0-08-044655-4.
- Ilchenko, N.O.; Sudarikov, D.V.; Slepukhin, P.A.; Rubtsova, S.A.; Kutchin, A.V. Synthesis of Chiral CF3-Contaning Pinane-Type Hydroxythiols. ChemistrySelect 2021, 6, 1710–1714.
- Banina, O.A.; Sudarikov, D.V.; Krymskaya, Y.V.; Frolova, L.L.; Kuchin, A.V. Synthesis of Chiral Hydroxythiols Based on Oxygen-Containing α- and β-Pinene Derivatives. Chem. Nat. Compd. 2015, 51, 261–265.
- Lezina, O.M.; Subbotina, S.N.; Frolova, L.L.; Rubtsova, S.A.; Sudarikov, D.V. Synthesis and Oxidative Transformations of New Chiral Pinane-Type γ-Ketothiols: Stereochemical Features of Reactions. Molecules 2021, 26, 5245.
- Gianini, M.; Zelewsky, A. von Synthesis of Chiral Thienylpyridines from Naturally Occurring Monoterpenes: Useful Ligands for Cyclometallated Complexes. Synthesis 1996, 1996, 702–706.
- Lu, C.-D.; Zakarian, A. Total Synthesis of (±)-Trichodermamide B and of a Putative Biosynthetic Precursor to Aspergillazine A Using an Oxaza-Cope Rearrangement. Angew. Chem. Int. Ed. 2008, 47, 6829–6831.
- Solladié, G.; Lohse, O. New Reagent for the Optical Resolution of Ketones: (−) (1R, 2R, 5R)-5-Methyl-2-(1-Mercapto-1-Methylethy)-Cyclohexanol. Application to Trans Dimethyl Cyclopentanone-3,4-Dicarboxylate. Tetrahedron Asymmetry 1993, 4, 1547–1552.
- Kawęcki, R. Facile Synthesis of Homochiral Derivatives of 10-Bornane Sulfinates, Sulfinamides and Sulfinimines. Tetrahedron Asymmetry 1999, 10, 4183–4190.
- Lynch, J.E. (4a R)-(4aα,7α,8aβ)-Hexahydro-4,4,7-Trimethyl-4 H -1,3-Benzoxathiin. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2001; p. rh014. ISBN 978-0-471-93623-7.
- Eliel, E.L.; Lynch, J.E.; Kume, F.; Frye, S.V. Chiral 1,3-Oxathiane from (+)-Pulegone: Hexahydro-4,4,7-Trimethyl-4h-1,3-Benzoxathiin. Org. Synth. 1987, 65, 215.
- Banina, O.A.; Sudarikov, D.V.; Slepukhin, P.A.; Frolova, L.L.; Kuchin, A.V. Stereoselective Synthesis of Carane-Type Hydroxythiols and Disulfides Based on Them. Chem. Nat. Compd. 2016, 52, 240–247.
- Paquette, L.A.; Ross, R.J.; Shi, Y.J. Regioselective Routes to Nucleophilic Optically Active 2- and 3-Carene Systems. J. Org. Chem. 1990, 55, 1589–1598.
- Nakano, T.; Shikisai, Y.; Okamoto, Y. Helix-Sense-Selective Free Radical Polymerization of 1-Phenyldibenzosuberyl Methacrylate. Polym. J. 1996, 28, 51–60.
- Taj, S.S.; Soman, R. Asymmetric Methylene Transfer Reactions I: Asymmetric Synthesis of Oxiranes from Carbonyl Compounds by Methylene Transfer Reaction Using Chiral S-Methyl-S-Neomenthyl-N-Tosyl Sulfoximines. Tetrahedron Asymmetry 1994, 5, 1513–1518.
- Mikołajczyk, M.; Perlikowska, W.; Omelańczuk, J. Synthesis of (+)-Neomenthanethiol and Some of Its Derivatives. A New Example of Asymmetric Induction in the Sulfoxide Synthesis. Synthesis 1987, 1987, 1009–1012.
- Banach, A.; Ścianowski, J.; Ozimek, P. The Use of Sulfides Derived from Carane, P-Menthane, Pinane, and Bornane in the Synthesis of Optically Active Epoxides. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 274–284.
- Izmest’ev, E.S.; Sudarikov, D.V.; Rubtsova, S.A.; Slepukhin, P.A.; Kuchin, A.V. Asymmetric Synthesis of New Optically Active Sulfinamides of Menthane Series and Their Derivatives. Russ. J. Org. Chem. 2012, 48, 184–192.
- Blanco, J.M.; Caamaño, O.; Eirín, A.; Fernández, F.; Medina, L. Synthesis of Chiral Sulfinic Acids: Sodium(1 S-Exo )-2-Bornanesulfinate. Synthesis 1990, 1990, 584–586.
- Li, J.; Ji, K.; Zheng, R.; Nelson, J.; Zhang, L. Expanding the Horizon of Intermolecular Trapping of in Situ Generated α-Oxo Gold Carbenes: Efficient Oxidative Union of Allylic Sulfides and Terminal Alkynes via C–C Bond Formation. Chem. Commun. 2014, 50, 4130–4133.
- Izmest’ev, E.S.; Sudarikov, D.V.; Rubtsova, S.A.; Slepukhin, P.A.; Kuchin, A.V. Synthesis and Reduction of New Sulfinimines with Isobornane Structure. Russ. J. Org. Chem. 2012, 48, 1407–1418.
- Adams, H.; Bell, R.; Cheung, Y.-Y.; Jones, D.N.; Tomkinson, N.C.O. The Cleavage of Meso-Epoxides with Homochiral Thiols: Synthesis of (+)- and (−)-Trans-1-Mercaptocyclohexan-2-Ol. Tetrahedron Asymmetry 1999, 10, 4129–4142.
- Aggarwal, V.K.; Kalomiri, M.; Thomas, A.P. Asymmetric Epoxidation Using Chiral Sulfur Ylides. Tetrahedron Asymmetry 1994, 5, 723–730.
- Rollin, P. Nucleophilic Inversions of a Chiral Alcohol Mediated by Zinc Salts. Synth. Commun. 1986, 16, 611–616.
- Aversa, M.C.; Barattucci, A.; Bonaccorsi, P.; Giannetto, P.; Nicolò, F. Structural Feature Relevance of the Chiral Alkyl Residue in the Addition Alkanesulfenic Acids/Enynes. Synthesis and Diels−Alder Reactivity of Enantiopure (E)-3-[(1S-Exo)-2-Bornylsulfinyl]-1- Methoxybuta-1,3-Dienes. J. Org. Chem. 1999, 64, 2114–2118.
- Hsu, J.-L.; Fang, J.-M. Triflate-Catalysed Addition of Thio Compounds to Camphene. J. Org. Chem. 2001, 66, 8573–8584.
- Uehara, K.; Tomooka, K. Planar Chiral Organosulfur Cycles. Chem. Lett. 2009, 38, 1028–1029.
- Galaka, T.; Ferrer Casal, M.; Storey, M.; Li, C.; Chao, M.; Szajnman, S.; Docampo, R.; Moreno, S.; Rodriguez, J. Antiparasitic Activity of Sulfur- and Fluorine-Containing Bisphosphonates against Trypanosomatids and Apicomplexan Parasites. Molecules 2017, 22, 82.
- Lee, S.M.; Cheng, Q.; Yu, X.; Liu, S.; Johnson, L.T.; Siegwart, D.J. A Systematic Study of Unsaturation in Lipid Nanoparticles Leads to Improved mRNA Transfection In Vivo. Angew. Chem. Int. Ed. 2021, 60, 5848–5853.
- Lee, D.-S.; Hung, S.-M.; Lai, M.-C.; Chu, H.-Y.; Yang, T.-K. Preparation of Optically Active New Mercapto Chiral Auxiliaries Derived from Camphor. Org. Prep. Proced. Int. 1993, 25, 673–679.
- Hung, S.-M.; Lee, D.-S.; Yang, T.-K. New Chiral Auxiliary: Optically Active Thiol Derived from Camphor. Tetrahedron Asymmetry 1990, 1, 873–876.
- Cocker, W.; Grayson, D.H. A Convenient Preparation of (−)-β-3,4-Epoxycarane. Tetrahedron Lett. 1969, 10, 4451–4452.
- Ishmuratov, G.Y.; Yakovleva, M.P.; Tukhvatshin, V.S.; Talipov, R.F.; Nikitina, L.E.; Artemova, N.P.; Startseva, V.A.; Tolstikov, A.G. Sulfur-Containing Derivatives of Mono- and Bicyclic Natural Monoterpenoids. Chem. Nat. Compd. 2014, 50, 22–47.
- Schoenauer, S.; Schieberle, P. Structure–Odor Activity Studies on Monoterpenoid Mercaptans Synthesized by Changing the Structural Motifs of the Key Food Odorant 1-p-Menthene-8-Thiol. J. Agric. Food Chem. 2016, 64, 3849–3861.
- Candela, K.; Fellous, R.; Joulain, D.; Faure, R. Reduction of Cis- and Trans-1,2-Epithio-p-Menth-8-Ene: Preparation of New Fragrant Terpenoid Thiols. Flavour Fragr. J. 2003, 18, 52–56.
- Blanco, J.M.; Caamano, O.; Fernandez, F.; Nieto, I. Syntheses of Chiral Menthyl and Neomenthyl Sulfides, sulfoxides and sulfones. J. Prakt. ChemieChemiker-Ztg. 1995, 337, 538–541.
- Izmest’ev, E.S.; Sudarikov, D.V.; Rubtsova, S.A.; Kuchin, A.V. Synthesis of Several Optically Active Menthane Disulfides and Thiosulfinates. Chem. Nat. Compd. 2011, 47, 46–50.
- Wilson, S.R.; Caldera, P.; Jester, M.A. Synthesis of Sulfides and Mercaptans from Thioketals. J. Org. Chem. 1982, 47, 3319–3321.
- Griffin, S.G.; Wyllie, S.G.; Markham, J.L.; Leach, D.N. The Role of Structure and Molecular Properties of Terpenoids in Determining Their Antimicrobial Activity. Flavour Fragr. J. 1999, 14, 322–332.
- Griffin, S.G.; Wyllie, S.G.; Markham, J.L. Antimicrobially Active Terpenes Cause K+ Leakage in E. coli Cells. J. Essent. Oil Res. 2005, 17, 686–690.
- Mancuso, M.; Catalfamo, M.; Laganà, P.; Rappazzo, A.C.; Raymo, V.; Zampino, D.; Zaccone, R. Screening of Antimicrobial Activity of Citrus Essential Oils against Pathogenic Bacteria and Candida Strains. Flavour Fragr. J. 2019, 34, 187–200.
- Spallek, M.J.; Storch, G.; Trapp, O. Straightforward Synthesis of Poly(Dimethylsiloxane) Phases with Immobilized (1R)-3-(Perfluoroalkanoyl)Camphorate Metal Complexes and Their Application in Enantioselective Complexation Gas Chromatography. Eur. J. Org. Chem. 2012, 2012, 3929–3945.
- Wu, H.-L.; Wu, P.-Y.; Cheng, Y.-N.; Uang, B.-J. Enantioselective Addition of Organozinc Reagents to Carbonyl Compounds Catalyzed by a Camphor Derived Chiral γ-Amino Thiol Ligand. Tetrahedron 2016, 72, 2656–2665.
- Wolinsky, J.; Marhenke, R.; Eustace, E.J. Sulfonation of Terpene Derivatives. Aluminum Hydride Desulfurization of Sultones. J. Org. Chem. 1973, 38, 1428–1430.
- Wolinsky, J.; Marhenke, R.L. Lithium Aluminum Hydride Reduction of Terpene Sultones. J. Org. Chem. 1975, 40, 1766–1770.
This entry is offline, you can click here to edit this entry!