Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Critical Care Medicine
Peptide nucleic acid (PNA) is an artificial DNA analog in which the negatively charged phosphodiester backbone is replaced by a charge-neutral pseudopeptide backbone. PNA exhibits several conformational flexibilities. It can adopt the A and B helical structures upon binding to target RNA and DNA, respectively, and form antiparallel and parallel duplexes. The antiparallel duplex is generally more stable than the parallel one.
- peptide nucleic acid
- PNA backbone
- base modification
- crosslinking
- therapeutic application
- genome editing
1. Outline of the Peptide Nucleic Acid Technology
PNA was first reported by Nielsen’s group in 1991 [8,77]. It comprises an electrically neutral aminoethyl glycine backbone. PNA is an artificial DNA analog in which the negatively charged phosphodiester backbone is replaced by a charge-neutral pseudopeptide backbone (Figure 1). PNA exhibits several conformational flexibilities. It can adopt the A and B helical structures upon binding to target RNA and DNA, respectively, and form antiparallel and parallel duplexes. The antiparallel duplex is generally more stable than the parallel one. The charge neutrality of PNA enabled its binding to the complementary DNA sequence target with increased affinity and sequence specificity, resulting in the unique mode of PNA action as follows. Considering the high affinity of PNA to DNA, single-stranded PNA (ssPNA) can invade dsDNA (Figure 1). Pseudocomplementary PNA (pcPNA) [78] was designed to form two PNA/DNA duplexes. pcPNA contains artificial nucleobases, where 2,6-diaminopurine (D) and 2-thiouracil (sU) are used instead of A and T, respectively, to avoid PNA/PNA self-duplex formation by the steric hindrance between them. The bifunctional PNA [79] and tail-clamp PNAs (tcPNAs) [80] were designed to “clamp” one DNA strand comprising two sections that are connected by a flexible linker that enables the invasion of the target DNA duplex. This invasion is initiated by the triplex formation of one section through Hoogsteen base pairing, and the other section forms a Watson–Crick base pairing with the same DNA strand, resulting in a PNA/DNA/PNA triplex. This “clamp” distorts the DNA structure, followed by the recruitment of endogenous repair factors that can be explored for genome editing. Further, tcPNA contains an extended Watson–Crick binding section to create a “tail” for distorting the target DNA for an extended stretch with increased affinity to DNA. The aforementioned PNA designs have their advantages, and their effectiveness has been demonstrated in antigene and genome editing.
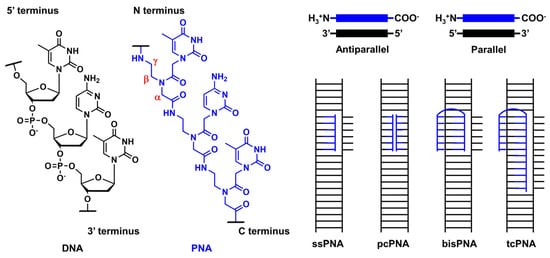
Figure 1. Structural features of PNA and unique mode of PNA actions.
2. Advancements in Peptic Nucleic Acid Engineering
The unique characteristics of PNA, the aforementioned charge neutrality, and structural flexibility, also cause several drawbacks, such as low water solubility, poor cellular uptake, self-aggregation, and orientational ambiguity, in target-sequence recognition. Many modifications have been explored to overcome these drawbacks to improve the application potential of PNAs. As many comprehensive and excellent reviews have described these modifications and applications [77,81,82,83,84], we only introduced selected examples, focusing on essential findings and recent advancements.
2.1. Modification of the Peptic Nucleic Acid Backbone
Ly’s group reported that the introduction of simple substituents, such as methyl, or hydroxymethyl, at the γ-position induced the preorganized, right-handed helical structure of the PNA backbone (Figure 2, 30), resulting in strengthened binding to complementary DNA and RNA [85]. The same group demonstrated that the modification of the γ-position of PNA with guanidine, γ-GPNA (Figure 2, 31), greatly improved the cellular uptake of γ-GPNA [86]. They also developed miniPEG γ-modified PNA (Figure 2, 32) exhibiting superior nucleic acid binding due to the better preorganization of its PNA backbone. The miniPEG γ-modified PNA can invade any dsDNA sequence through only Watson–Crick base pairing to recognize the target [87]. Tahtinen’s group developed γ-guanidinylmethyl PNA (Figure 2, 33) for recognizing triplex-forming PNAs. Three consecutive incorporations of 33 in PNA achieved the best binding affinity and Hoogsteen-face selectivity of the oligomer with improved cellular uptake [88]. Presently, the aforementioned γ-GPNA and miniPEG γ-modified PNA are widely used PNA derivatives for therapeutic purposes.
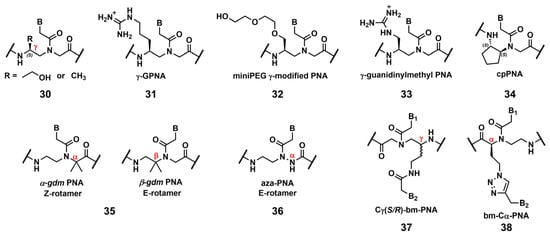
Figure 2. Chemical modifications of the PNA backbone. The position of the carbon is mentioned using α, β, γ (red).
Appella’s group reported the detailed biophysical and structural properties of S,S-cyclopentyl PNA, cpPNA (Figure 2, 34) [89,90]. The cyclopentane ring restricts the conformational flexibility of the PNA backbone, thus inducing a right-handed helix that favors binding to complementary DNA. Further, the affinity and selectivity improved with an increased amount of 34, which enabled the customization of the stability of the complex. Recently, Ganesh’s group reported that the introduction of the gem-dimethyl (gdm) group influenced the Z/E rotamer ratio of the tertiary amide. The α-gdm monomer exclusively exhibits the Z-rotamer, whereas the β-gdm monomer exhibits the E-rotamer (Figure 2, 35) [91,92]. Those E/Z-rotamers influenced the orientation preference of PNA in the formation of the complex. The same research group also reported aza-PNA bearing a nitrogen atom instead of a carbon atom at the α-position (Figure 2, 36) [93]. Interestingly, the aza-PNA monomer assumed the E-form via an eight-membered hydrogen-bonded ring with backbone folding. A future study will discuss how this modification impacts its target recognition.
A novel class of PNAs, bimodal PNA, Cγ(S/R)-bm-PNA (Figure 2, 37), was developed by Ganesh’s group [94,95,96,97]. Cγ(S/R)-bm-PNA contains an additional nucleobase at the γ-position of the PNA backbone, which enables bifacial recognition-forming duplexes at the B1 and B2 sides. The thermal stability of the DNA1/Cγ(S/R)-bm-PNA/DNA2 complexes was higher than those of their respective isolated duplexes [94,95]. The other bimodal PNA, bm-Cα-PNA (Figure 2, 38), contains an additional nucleobase at the α-position of the PNA backbone [96]. Additionally, 38, and 37 exhibited similar properties, although the target sequences were restricted to homothymine and homocytosine. These bimodal PNAs can be used to generate novel higher-order assemblies with DNAs and RNAs. For example, they reported a pentameric complex comprising a triplex and two duplexes, DNA1–Cγ(S/R)-bm-PNA/DNA2–Cγ(S/R)-bm-PNA/DNA3 [97]. Further investigations of these new bimodal PNAs, as well as their biological applications, are anticipated.
2.2. Base Modification of Peptide Nucleic Acid
2.2.1. Base Modifications for Enhanced Triplex Formation
PNA forms a triplex structure via the Hoogsteen hydrogen-bond-forming T·A:T triad and C+·G:C triad base pairing. Therefore, the inability to form stable hydrogen bonds with the T:A and C:G base pairs, as well as the necessity of protonating C, limit their in vivo application, including parallel TFOs. Some of the designed base analogs for TFOs can be used for PNA. For example, the base analogs 2 [98] and 5 [99] (Figure 3) can be used for T:A and C:G base-pair recognition, and 5-mC, pseudoisocytosine (Figure 3, 12) [100] can be used to replace C. Several artificial bases were originally developed for PNA [101,102]. Recently, Rozner’s group systematically surveyed simple nitrogen heterocycles for C:G base-pair recognition and observed that 3-pyridazinyl nucleobase, PN (Figure 4, 39), forms more stable hydrogen bonds than the other heterocycles [102].
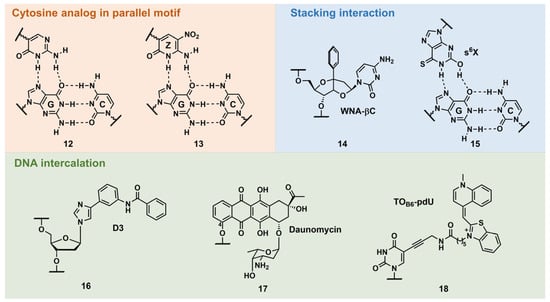
Figure 3. Selected examples of other types of triplex-stabilizing artificial nucleobases.
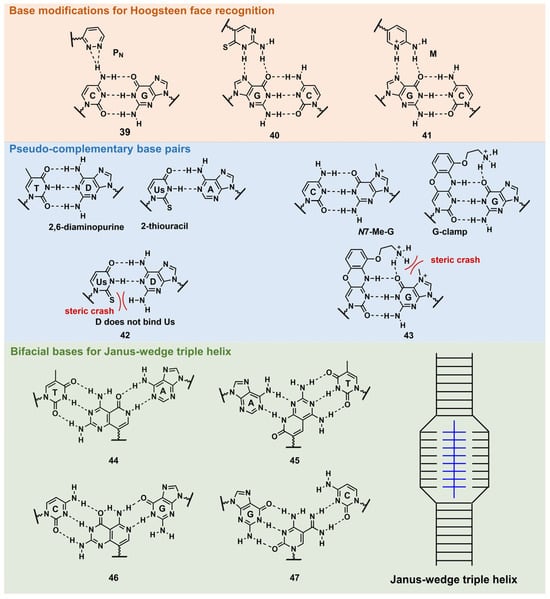
Figure 4. Nucleobase modifications of PNA.
Thio-pseudoisocytosine (Figure 4, 40) [103] was studied by Chen’s group as a replacement for C. Here, 40 forms stable base pairing through the synergistic effect of improved van der Waals contacts base stacking with hydrogen bond formation. Rozner’s group examined 2-aminopyridine M (Figure 4, 41) as a more basic C nucleobase [104,105]. The replacement of six pseudoisocytosines in 9-mer PNA comprising six Ms increased the affinity of PNA to dsDNA by ≈100-fold owing to the cationic character of M.
2.2.2. Base Modifications for Peptide Nucleic Acid Functionalization
As mentioned in Figure 1, D and sU were designed to avoid the formation of an unproductive PNA–PNA duplex for pcPNA via a steric clash between the 2-amino group of D and thiocarbonyl group of sU (Figure 4, 42) [106]. Hudson’s group reported an improved synthesis of sU, which eases the preparation of pcPNA and will accelerate future pcPNA studies [107]. Recently, Winssinger’s group reported a new pseudocomplementary G:C base pair for G-clamp-based dsDNA invasion (Figure 4, 43) [108].
G-clamp is a phenoxazine-derived tricyclic C analog that can strongly bind to G through additional interactions, such as π-stacking, the electrostatic attraction of a positively charged amine, and hydrogen bonding at the Hoogsteen face [109]. The introduction of G-clamp into PNA significantly improved the thermodynamic stability of the PNA/DNA duplex [110]. They developed N-7 methylguanine (N7-Me-G), which was designed to cause steric and electrostatic repulsions between the G-clamp. The modified PNAs were used in the detection of the dsDNA target, the RT-RPA amplicon, from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), with single nucleotide resolution, discriminating between two SARS-CoV-2 strains. Each strand of modified PNA exhibited fast strand invasion in the physiological condition and formed stable complexes with low equivalents of PNA.
An investigation of the Janus–Wedge triple PNA helix was pioneered by McLaughlin’s group [111]. Therein, bifacial artificial PNA nucleobases form hydrogen bonds with the Watson–Crick faces of the two DNA target strands. In 2018, Thadke’s group first reported a complete set of bifacial nucleobases that can distinguish the T:A, A:T, C:G, and G:C base pairs (Figure 4, 44–47) [112,113]. The 6-mer miniPEG γ-modified PNA comprising these nucleobases efficiently and rapidly invaded target dsDNA with high sequence specificity under physiological conditions [113]. As 44–47 enable the targeting of any sequences, they could be novel gene-targeting tools, and their applications for therapeutic purposes will be investigated subsequently.
2.3. Recent Advancements in Crosslinkable Peptide Nucleic Acid for DNA Targeting
Glazer and Nielsen’s group developed psoralen-conjugated pcPNAs, demonstrating that they can be used to induce single-base substitutions and deletions within the target site [114,115]. The linker was only linked to the 8-position of psoralen in these PNAs, making the effect of the linker position on crosslinking formation elusive. Recently, our group developed novel psoralen NHS esters (Figure 5, 21 and 22), which enabled the easy conjugation of psoralen in the N-terminus of PNA and subsequent photo-crosslinking evaluation [57]. The yields of the adduct products of 5-Ps–PNA and trioxsalen–PNA prepared with 21 and 22 were 48% and 45%, respectively, after 30 s of irradiation; the yields were much higher than those of corresponding PNAs prepared using SPB (15%). The applications of these novel Ps–PNAs are being studied in our lab.
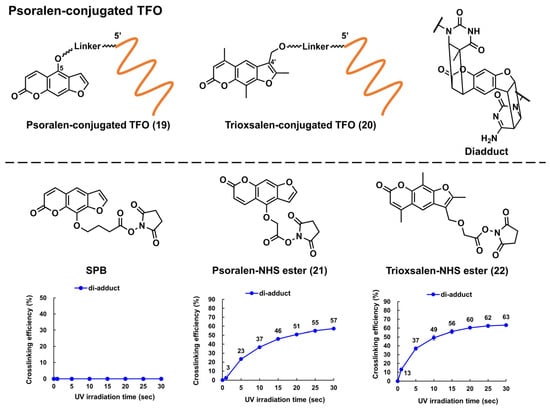
Figure 5. Structures of the psoralen-conjugated TFOs (Ps–TFOs) and psoralen N-hydroxysuccinimide (NHS) esters. The charts show the crosslinking efficiencies of the corresponding Ps–TFOs prepared using each NHS ester (the charts were quoted from [57]).
Crosslinkable furan-derived nucleobases were developed for PNA. The furan generates reactive species immediately after the activation of reactive oxygen species (ROS), which can be exploited for DNA crosslinking. Sequence-specific crosslinking was realized with γ-Lys-modified PNA [116,117]. Recently, Vilaivan’s group reported novel pyrrolidinyl PNA probes, exhibiting furan as a crosslinking formation with target DNA. The positional effect on crosslinking was examined, revealing that the external incorporation of the furan moiety improved the crosslinking. A future study will discuss its biological applications [118].
3. Recent Advancements in the Therapeutic Applications of Peptide Nucleic Acids
3.1. Modulation of DNA Expression
Recently, Tonelli’s group reported the therapeutic applications of PNAs in targeting the MYCN gene [119,120]. The PNA employed in these studies was named BGA002, which is conjugated with a nuclear localization signal peptide [87] and specific to the MYCN gene. Although the sequence information was not disclosed, BGA002 exhibited more potent biological activities than their previous MYCN-targeting PNA (BGA001), which also exhibited antitumor activity in mice with rhabdomyosarcomas [121]. With PNA BGA002, they targeted MYCN-amplified neuroblastoma (MNA–NB) cells, in which MYCN was associated with increased ROS, downregulated mitophagy, and a poor prognosis. BGA002 inhibited the expression of the MYCN gene, causing profound mitochondrial damage through the downregulation of the mitochondrial molecular chaperone TRAP1; ROS increased with the concomitant decrease in the MNA–NB xenograft tumor. This research first described the relevance of MYCN in MNA–NB in mitochondrial maintenance [119]. In other research [120], the combinational use of BGA002 and retinoic acid (RA) was found to be beneficial in the treatment of MNA–NB. RA has been used to treat MNA–NB patients. However, RA resistance was observed for some patients. The coadministration of BGA002 and RA mediated the therapeutic efficacy of RA by inhibiting BGA002 on the MYCN gene. The inhibition of the MYCN gene by BGA002 decreased the mTOR pathway activity, followed by the autophagy response of MNA–NB. The efficacy of BGA002 treatment with RA was also demonstrated in an MNA–NB mouse model.
3.2. Genome Editing
In 2016, Glazer et al. reported an in vivo correction of a β-thalassemia mutation in mice by miniPEG γ-modified tcPNA (γtcPNA) [122]. An γtcPNA and donor-DNA injection with poly(lacto-glycolic acid) (PLGA) NPs, as well as a stem-cell-factor treatment, ameliorated the disease phenotype and collected the β-globin gene by up to 7% in hematopoietic stem cells. In utero experiments have also been performed using the same mouse model, achieving improvements in the disease phenotype in pups [123]. It seems that the tcPNA works by opening the target dsDNA via strand invasion (Figure 1), which permits the donor DNA to hybridize the target DNA. Finally, the homology-directed repair using this donor DNA as a template resulted in gene collection [124]. In 2022, Piotrowski-Daspit’s group further demonstrated the utility of PLGA NPs encapsulating PNA miniPEG γ-modified tcPNA and donor DNAs in cystic fibrosis (CF) treatment. CF patients experience multiorgan dysfunction, which is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. The in vivo correction of the CF mouse model using γtcPNA resulted in a partial gain of CFTR function and improved the phenotype [125]. Recently, Glazer’s group also applied the same system to genome editing in single-cell embryos containing mutated eGFP genes. The blastocysts from the embryos that were treated with γtcPNA exhibited the expression of corrected eGFP with high editing levels, up to 94%. The mice from the re-implanted embryos consistently exhibited editing. This research is the first example of embryonic-gene editing using PNA [126]. In these above experiments, gene editing was very site-specific, with considerably low levels of off-target sequence modification.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics15102515
This entry is offline, you can click here to edit this entry!