The landscape of diagnosing and treating endometrial cancer is undergoing a profound transformation due to the integration of molecular analysis and innovative therapeutic approaches. Tailoring treatments based on specific biomarkers has evolved into a standard practice in both initial and recurrent therapy protocols. Diverse biological abnormal changes in pathways have been discerned in EC cells. This has prompted the active development of novel therapeutic drugs and biomarkers, including immunomodulation inhibitors targeting programmed cell death protein 1 (PD-1) or programmed cell death ligand 1 (PD-L1), to address these anomalies.
- endometrial cancer
- PD-1
- PD-L1
- molecular classification
- immune-checkpoint inhibitor
1. Introduction
2. Immune Micro-Environment in Endometrial Cancer
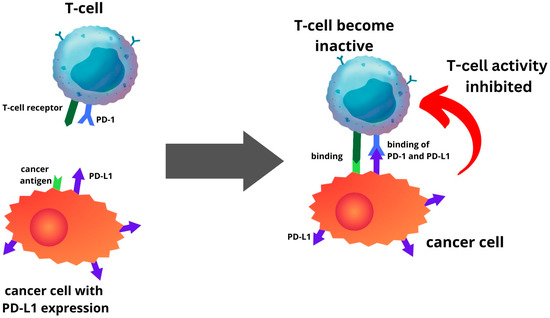
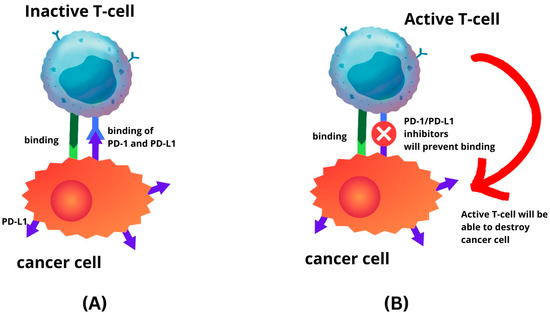
3. Expression of PD-1 and PD-L1 in Endometrial Cancer
This entry is adapted from the peer-reviewed paper 10.3390/ijms242015233
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249.
- Gentry-Maharaj, A.; Karpinskyj, C. Current and Future Approaches to Screening for Endometrial Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 65, 79–97.
- Thomas, S.; Hussein, Y.; Bandyopadhyay, S.; Cote, M.; Hassan, O.; Abdulfatah, E.; Alosh, B.; Guan, H.; Soslow, R.A.; Ali-Fehmi, R. Interobserver Variability in the Diagnosis of Uterine High-Grade Endometrioid Carcinoma. Arch. Pathol. Lab. Med. 2016, 140, 836–843.
- Gilks, C.B.; Oliva, E.; Soslow, R.A. Poor Interobserver Reproducibility in the Diagnosis of High-Grade Endometrial Carcinoma. Am. J. Surg. Pathol. 2013, 37, 874–881.
- de Boer, S.M.; Wortman, B.G.; Bosse, T.; Powell, M.E.; Singh, N.; Hollema, H.; Wilson, G.; Chowdhury, M.N.; Mileshkin, L.; Pyman, J.; et al. Clinical Consequences of Upfront Pathology Review in the Randomised PORTEC-3 Trial for High-Risk Endometrial Cancer. Ann. Oncol. 2018, 29, 424–430.
- Golia D’Augè, T.; Cuccu, I.; Santangelo, G.; Muzii, L.; Giannini, A.; Bogani, G.; Di Donato, V. Novel Insights into Molecular Mechanisms of Endometrial Diseases. Biomolecules 2023, 13, 499.
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73.
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP Guidelines for the Management of Patients with Endometrial Carcinoma. Int. J. Gynecol. Cancer 2021, 31, 12–39.
- Vanderstraeten, A.; Tuyaerts, S.; Amant, F. The Immune System in the Normal Endometrium and Implications for Endometrial Cancer Development. J. Reprod. Immunol. 2015, 109, 7–16.
- Ochiel, D.O.; Ghosh, M.; Fahey, J.V.; Guyre, P.M.; Wira, C.R. Human Uterine Epithelial Cell Secretions Regulate Dendritic Cell Differentiation and Responses to TLR Ligands. J. Leukoc. Biol. 2010, 88, 435–444.
- Agostinis, C.; Mangogna, A.; Bossi, F.; Ricci, G.; Kishore, U.; Bulla, R. Uterine Immunity and Microbiota: A Shifting Paradigm. Front. Immunol. 2019, 10, 2387.
- Wira, C.R.; Fahey, J.V.; Rodriguez-Garcia, M.; Shen, Z.; Patel, M.V. Regulation of Mucosal Immunity in the Female Reproductive Tract: The Role of Sex Hormones in Immune Protection against Sexually Transmitted Pathogens. Am. J. Reprod. Immunol. 2014, 72, 236–258.
- Wang, F.; Qualls, A.E.; Marques-Fernandez, L.; Colucci, F. Biology and Pathology of the Uterine Microenvironment and Its Natural Killer Cells. Cell. Mol. Immunol. 2021, 18, 2101–2113.
- Aflatoonian, R.; Amjadi, F.; Mehdizadeh, M.; Salehi, E. Role of the Innate Immunity in Female Reproductive Tract. Adv. Biomed. Res. 2014, 3, 1.
- Oertelt-Prigione, S. Immunology and the Menstrual Cycle. Autoimmun. Rev. 2012, 11, A486–A492.
- Zwahlen, M.; Stute, P. Impact of Progesterone on the Immune System in Women: A Systematic Literature Review. Arch. Gynecol. Obstet. 2023, 1–10.
- Morelli, S.; Mandal, M.; Goldsmith, L.T.; Kashani, B.N.; Ponzio, N.M. The Maternal Immune System during Pregnancy and Its Influence on Fetal Development. Res. Rep. Biol. 2015, 6, 171–189.
- Chen, R.Y.; Zhu, Y.; Shen, Y.Y.; Xu, Q.Y.; Tang, H.Y.; Cui, N.X.; Jiang, L.; Dai, X.M.; Chen, W.Q.; Lin, Q.; et al. The Role of PD-1 Signaling in Health and Immune-Related Diseases. Front. Immunol. 2023, 14, 1163633.
- Chikuma, S. Basics of PD-1 in Self-Tolerance, Infection, and Cancer Immunity. Int. J. Clin. Oncol. 2016, 21, 448–455.
- Laba, S.; Mallett, G.; Amarnath, S. The Depths of PD-1 Function within the Tumor Microenvironment beyond CD8+ T Cells. Semin. Cancer Biol. 2022, 86, 1045–1055.
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 2020, 11, 487.
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998.
- Burnet, M. Cancer-A Biological Approach* Iii. Viruses Associated with Neoplastic Conditions. Br. Med. J. 1957, 1, 841.
- Stutman, O. Tumor Development after 3-Methylcholanthrene in Immunologically Deficient Athymic-Nude Mice. Science 1974, 183, 534–536.
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The Roles of IFNγ in Protection against Tumor Development and Cancer Immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109.
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167.
- Morrison, J.; Balega, J.; Buckley, L.; Clamp, A.; Crosbie, E.; Drew, Y.; Durrant, L.; Forrest, J.; Fotopoulou, C.; Gajjar, K.; et al. British Gynaecological Cancer Society (BGCS) Uterine Cancer Guidelines: Recommendations for Practice. Eur. J. Obstet. Gynecol. Reprod. Biol. 2022, 270, 50–89.
- Mellman, I.; Steinman, R.M. Dendritic Cells: Specialized and Regulated Antigen Processing Machines. Cell 2001, 106, 255–258.
- Palucka, K.; Banchereau, J.; Mellman, I. Designing Vaccines Based on Biology of Human Dendritic Cell Subsets. Immunity 2010, 33, 464–478.
- Chimal-Ramírez, G.K.; Espinoza-Sánchez, N.A.; Fuentes-Pananá, E.M. Protumor Activities of the Immune Response: Insights in the Mechanisms of Immunological Shift, Oncotraining, and Oncopromotion. J. Oncol. 2013, 2013, 835956.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570.
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the Equilibrium in Cancer Immunoediting: From Tumor Tolerance to Eradication. Immunol. Rev. 2011, 241, 104–118.
- Aguirre-Ghiso, J.A. Models, Mechanisms and Clinical Evidence for Cancer Dormancy. Nat. Rev. Cancer 2007, 7, 834–846.
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive Immunity Maintains Occult Cancer in an Equilibrium State. Nature 2007, 450, 903–907.
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages in Vivo. Blood 1998, 92, 4150–4166.
- Yoshimura, A.; Muto, G. TGF-β Function in Immune Suppression. In Negative Co-Receptors and Ligands; Springer: Berlin/Heidelberg, Germany, 2010; Volume 350, pp. 127–147.
- Löb, S.; Königsrainer, A.; Rammensee, H.G.; Opelz, G.; Terness, P. Inhibitors of Indoleamine-2,3-Dioxygenase for Cancer Therapy: Can We See the Wood for the Trees? Nat. Rev. Cancer 2009, 9, 445–452.
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Pérez-Villar, J.J.; López-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for Immunosurveillance of Altered HLA Class I Phenotypes in Human Tumours. Immunol. Today 1997, 18, 89–95.
- Ventriglia, J.; Paciolla, I.; Pisano, C.; Cecere, S.C.; Di Napoli, M.; Tambaro, R.; Califano, D.; Losito, S.; Scognamiglio, G.; Setola, S.V.; et al. Immunotherapy in Ovarian, Endometrial and Cervical Cancer: State of the Art and Future Perspectives. Cancer Treat. Rev. 2017, 59, 109–116.
- Bretscher, P.A. A Two-Step, Two-Signal Model for the Primary Activation of Precursor Helper T Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 185–190.
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The Future of Cancer Treatment: Immunomodulation, CARs and Combination Immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290.
- Appleman, L.J.; Boussiotis, V.A. T Cell Anergy and Costimulation. Immunol. Rev. 2003, 192, 161–180.
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61.
- Townsend, S.E.; Allison, J.P. Tumor Rejection after Direct Costimulation of CD8+ T Cells by B7-Transfected Melanoma Cells. Science 1993, 259, 368–370.
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086.
- Linsley, P.S.; Greene, J.A.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) Bind with Similar Avidities but Distinct Kinetics to CD28 and CTLA-4 Receptors. Immunity 1994, 1, 793–801.
- Linsley, P.S.; Brady, W.; Urnes, M.; Grosmaire, L.S.; Damle, N.K.; Ledbetter, J.A. CTLA4 Is a Second Receptor for the b Cell Activation Antigen B7. J. Exp. Med. 1991, 174, 561–569.
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217.
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and Are Functionally Impaired. Blood 2009, 114, 1537–1544.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Simon, S.; Labarriere, N. PD-1 Expression on Tumor-Specific T Cells: Friend or Foe for Immunotherapy? OncoImmunology 2018, 7, e1364828.
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814.
- Li, C.; Li, W.; Xiao, J.; Jiao, S.; Teng, F.; Xue, S.; Zhang, C.; Sheng, C.; Leng, Q.; Rudd, C.E.; et al. ADAP and SKAP 55 Deficiency Suppresses PD-1 Expression in CD 8 + Cytotoxic T Lymphocytes for Enhanced Anti-tumor Immunotherapy. EMBO Mol. Med. 2015, 7, 754–769.
- Salmaninejad, A.; Khoramshahi, V.; Azani, A.; Soltaninejad, E.; Aslani, S.; Zamani, M.R.; Zal, M.; Nesaei, A.; Hosseini, S.M. PD-1 and Cancer: Molecular Mechanisms and Polymorphisms. Immunogenetics 2018, 70, 73–86.
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 455837.
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The Function of Programmed Cell Death 1 and Its Ligands in Regulating Autoimmunity and Infection. Nat. Immunol. 2007, 8, 239–245.
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human Cancer Immunotherapy with Antibodies to the PD-1 and PD-L1 Pathway. Trends Mol. Med. 2015, 21, 24–33.
- Ji, M.; Liu, Y.; Li, Q.; Li, X.D.; Zhao, W.Q.; Zhang, H.; Zhang, X.; Jiang, J.T.; Wu, C.P. PD-1/PD-L1 Pathway in Non-Small-Cell Lung Cancer and Its Relation with EGFR Mutation. J. Transl. Med. 2015, 13, 5.
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from Lymphocytes Induces PD-L1 Expression and Promotes Progression of Ovarian Cancer. Br. J. Cancer 2015, 112, 1501–1509.
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-Induced Activation of JAK1 and JAK2 Suppresses Tumor Cell Susceptibility to NK Cells through Upregulation of PD-L1 Expression. Oncoimmunology 2015, 4, e1008824.
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201.
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386.
- Nunes-Xavier, C.E.; Angulo, J.C.; Pulido, R.; López, J.I. A Critical Insight into the Clinical Translation of PD-1/PD-L1 Blockade Therapy in Clear Cell Renal Cell Carcinoma. Curr. Urol. Rep. 2019, 20, 1.
- Cao, W.; Ma, X.; Fischer, J.V.; Sun, C.; Kong, B.; Zhang, Q. Immunotherapy in Endometrial Cancer: Rationale, Practice and Perspectives. Biomark. Res. 2021, 9, 49.
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856.
- Mamat @ Yusof, M.N.; Chew, K.T.; Hafizz, A.M.H.A.; Abd Azman, S.H.; Ab Razak, W.S.; Hamizan, M.R.; Kampan, N.C.; Shafiee, M.N. Efficacy and Safety of PD-1/PD-L1 Inhibitor as Single-Agent Immunotherapy in Endometrial Cancer: A Systematic Review and Meta-Analysis. Cancers 2023, 15, 4032.
- Heinzerling, L.; Kirchberger, M.C.; Walter, L.; Schuler, G. Predicting the Response to Anti-PD1 Therapy in Metastatic Melanoma. Transl. Cancer Res. 2016, 5, S576–S579.
- Chen, S.; Zhang, Z.; Zheng, X.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Cui, P.; et al. Response Efficacy of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 562315.
- Adkins, D.R.; Haddad, R.I. Clinical Trial Data of Anti–PD-1/PD-L1 Therapy for Recurrent or Metastatic Nasopharyngeal Carcinoma: A Review. Cancer Treat. Rev. 2022, 109, 102428.
- Crumley, S.; Kurnit, K.; Hudgens, C.; Fellman, B.; Tetzlaff, M.T.; Broaddus, R. Identification of a Subset of Microsatellite-Stable Endometrial Carcinoma with High PD-L1 and CD8+ Lymphocytes. Mod. Pathol. 2019, 32, 396–404.
- Mamat @ Yusof, M.N.; Chew, K.T.; Kampan, N.; Nor, N.H.; Md Zin, R.R.; Tan, G.C.; Shafiee, M.N. PD-L1 Expression in Endometrial Cancer and Its Association with Clinicopathological Features: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 3911.
- Zhang, S.; Minaguchi, T.; Xu, C.; Qi, N.; Itagaki, H.; Shikama, A.; Tasaka, N.; Akiyama, A.; Sakurai, M.; Ochi, H.; et al. PD-L1 and CD4 Are Independent Prognostic Factors for Overall Survival in Endometrial Carcinomas. BMC Cancer 2020, 20, 127.
- Sungu, N.; Yildirim, M.; Desdicioglu, R.; Aydoǧdu, Ö.B.; Kiliçarslan, A.; Doǧan, H.T.; Yazgan, A.K.; Akyol, M.; Erdoǧan, F. Expression of Immunomodulatory Molecules PD-1, PD-L1, and PD-L2, and Their Relationship with Clinicopathologic Characteristics in Endometrial Cancer. Int. J. Gynecol. Pathol. 2019, 38, 404–413.
- Engerud, H.; Berg, H.F.; Myrvold, M.; Halle, M.K.; Bjorge, L.; Haldorsen, I.S.; Hoivik, E.A.; Trovik, J.; Krakstad, C. High Degree of Heterogeneity of PD-L1 and PD-1 from Primary to Metastatic Endometrial Cancer. Gynecol. Oncol. 2020, 157, 260–267.
- Chew, M.; Wong, Y.P.; Karim, N.; Mustangin, M.; Alfian, N.; Tan, G.C. Programmed Death Ligand 1: A Poor Prognostic Marker in Endometrial Carcinoma. Diagnostics 2020, 10, 394.
- Mo, Z.; Liu, J.; Zhang, Q.; Chen, Z.; Mei, J.; Liu, L.; Yang, S.; Li, H.; Zhou, L.; You, Z. Expression of PD-1, PD-L1 and PD-L2 Is Associated with Differentiation Status and Histological Type of Endometrial Cancer. Oncol. Lett. 2016, 12, 944–950.
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase E-Mutated and Microsatellite-Instable Endometrial Cancers with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323.
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship with PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-Based Approach. Ann. Oncol. 2019, 30, 1232–1243.
- Vanderwalde, A.; Spetzler, D.; Xiao, N.; Gatalica, Z.; Marshall, J. Microsatellite Instability Status Determined by Next-Generation Sequencing and Compared with PD-L1 and Tumor Mutational Burden in 11,348 Patients. Cancer Med. 2018, 7, 746–756.
- Pasanen, A.; Ahvenainen, T.; Pellinen, T.; Vahteristo, P.; Loukovaara, M.; Bützow, R. PD-L1 Expression in Endometrial Carcinoma Cells and Intratumoral Immune Cells: Differences across Histologic and TCGA-Based Molecular Subgroups. Am. J. Surg. Pathol. 2020, 44, 174–181.
- Li, Z.; Joehlin-Price, A.S.; Rhoades, J.; Ayoola-Adeola, M.; Miller, K.; Parwani, A.V.; Backes, F.J.; Felix, A.S.; Suarez, A.A. Programmed Death Ligand 1 Expression among 700 Consecutive Endometrial Cancers: Strong Association with Mismatch Repair Protein Deficiency. Int. J. Gynecol. Cancer 2018, 28, 59–68.
- Zong, L.; Sun, Z.; Mo, S.; Lu, Z.; Yu, S.; Xiang, Y.; Chen, J. PD-L1 Expression in Tumor Cells Is Associated with a Favorable Prognosis in Patients with High-Risk Endometrial Cancer. Gynecol. Oncol. 2021, 162, 631–637.
- Asaka, S.; Yen, T.T.; Wang, T.L.; Shih, I.M.; Gaillard, S. T Cell-Inflamed Phenotype and Increased Foxp3 Expression in Infiltrating T-Cells of Mismatch-Repair Deficient Endometrial Cancers. Mod. Pathol. 2019, 32, 576–584.
- Talhouk, A.; Derocher, H.; Schmidt, P.; Leung, S.; Milne, K.; Blake Gilks, C.; Anglesio, M.S.; Nelson, B.H.; McAlpine, J.N. Molecular Subtype Not Immune Response Drives Outcomes in Endometrial Carcinoma. Clin. Cancer Res. 2019, 25, 2537–2548.
- Bregar, A.; Deshpande, A.; Grange, C.; Zi, T.; Stall, J.; Hirsch, H.; Reeves, J.; Sathyanarayanan, S.; Growdon, W.B.; Rueda, B.R. Characterization of Immune Regulatory Molecules B7-H4 and PD-L1 in Low and High Grade Endometrial Tumors. Gynecol. Oncol. 2017, 145, 446–452.