1. Introduction
Endometrial cancer (EC) is the sixth most common malignancy among women worldwide [
1]. Geographically, EC is more common in the Northern and Southern Hemispheres, Europe, and Oceania/Australasia, while it is less common in Africa and South and Central Asia. The most common causes of EC include a family history of the disease, menstrual abnormalities, infertility, exposure to oestrogen, the use of hormonal drugs, obesity, diabetes, and a high body mass index [
2]. Pathological data, including histology, stage, grade, myometrial invasion, lymph vascular space invasion, and cervical stromal invasion, have been used for decades to determine the recurrence risk of EC. However, expert gynaecologic pathologists may face challenges in classifying the risk based on pathological findings [
3,
4]. Histology, grade, stage, and risk classification criteria, such as cervical stromal invasion, myometrial invasion, and lymph vascular space invasion varied among reviewers in past therapeutic studies that required central pathology evaluation [
5]. As a result, some patients may not receive appropriate care or treatment due to inconsistent risk categorization.
Recent advancements in genome analysis technology have revealed genomic anomalies within EC. Additionally, integrated genomic analyses have identified molecular subgroups that align with prognosis. The most notable approach of integrating molecular characteristics with EC classification by the Cancer Genome Atlas (TCGA) has resolved the numerous limitations in risk stratification. Later, a new model named ProMisE (Proactive Molecular Risk Classifier for Endometrial Cancer) was introduced to improve the limitation of the TCGA methodologies used for immediate clinical application. Several studies showed that this paradigm is effective for diagnostic specimens like endometrial biopsies, curettages, and final hysterectomy specimens. This model was implemented in the European Society of Gynecological Oncology (ESGO), the European Society for Radiotherapy and Oncology (ESTRO), and the European Society of Pathology (ESP) 2020 Guidelines for the management of EC patients based on tumour aggressiveness and recurrence [
6,
7,
8]. These findings are expected to play a significant role in guiding treatment and management decisions for EC [
7,
8]. Furthermore, diverse biological abnormal changes in pathways have been discerned in EC cells. This has prompted the active development of novel therapeutic drugs and biomarkers, including immunomodulation inhibitors targeting programmed cell death protein 1 (PD-1) or programmed cell death ligand 1 (PD-L1), to address these anomalies.
2. Immune Micro-Environment in Endometrial Cancer
The immune system plays a multifaceted role in the normal endometrium, particularly within endometrial epithelial cells. Acting as a part of the mucosal immune system, it serves as a physical barrier and is involved in the production of defensins, other immune mediators, and antigens [
25,
26,
27,
28]. In a normal endometrium, both the innate and adaptive immune systems are pivotal in eliminating pathogens, generating inflammatory cytokines, and regulating immune responses [
25,
29,
30]. These functions are intricately regulated by sex hormones, particularly oestradiol and progesterone, which exhibit variations corresponding to the fluctuations in the menstrual cycle [
25,
31,
32]. The immune system of the endometrium comprises various components, all with the dual purpose of maintaining normal physiological processes. It creates an environment that suppresses immune responses to prevent the mother’s immune system from rejecting the foetus, while simultaneously protecting the compromised endometrium from potential infections during menstruation [
25,
33]. Under normal circumstances, PD-1 serves to inhibit autoimmunity, restrict damage from infections to healthy tissues, and promote self-tolerance [
34,
35]. Elevated PD-1 expression on T cells can impact their ability to combat cancer and infectious diseases [
34,
36,
37].
A cancerous endometrium exhibits a distinctive microenvironment where carcinogenic substances can directly impact immune-related signalling pathways or the host’s defensive inflammation, leading to changes in immunological balance through tumour-induced immunoediting. Throughout the process of carcinogenesis, the endometrial immune response can both promote and inhibit tumour growth. The “cancer immunosurveillance” hypothesis, initially proposed by Burnet and Thomas in the late 1950s, posited that tumours in immunocompetent hosts trigger an immune system response aimed at restricting malignant cell proliferation [
38,
39]. This conceptual framework aimed to connect the immune system with tumour progression. However, subsequent experimental findings contradicted this hypothesis, leading to ongoing debates. Consequently, the cancer immunosurveillance concept fell out of favour [
40]. In 2002, Ikeda, Old, and Schreiber introduced a more intricate model known as “Cancer Immunoediting”, suggesting that the immune system can simultaneously impede and accelerate tumour growth [
38,
41]. This model comprises three phases: elimination, equilibrium, and escape. This traditional model will be employed to illustrate the reciprocal interactions between EC tumours and immunity. During the elimination phase, both innate and adaptive immune responses identify and eliminate EC cells through cytotoxic mechanisms [
42]. Dendritic cells (DCs) play a role by phagocytosing and processing “altered self” EC cells and “non-self” antigens under conditions of stress and harm [
43,
44]. DCs then stimulate and present tumour-associated antigens to generate CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ T cells [
44,
45]. CD8+ CTLs directly destroy EC cells, while CD4+ T helper cells activate specific B cell responses for both humoral and cytotoxic immune reactions [
46]. The cancer immunoediting process concludes when the immune system successfully eliminates all EC cells [
47]. Despite this elimination phase, a small fraction of malignant cells may survive and enter a state of equilibrium. During the “equilibrium” phase, EC cells and the immune system establish a dynamic balance, resulting in a period of temporal biological equilibrium [
48]. Numerous dormant EC cells can remain latent in patients for extended periods [
49]. This period is characterised by complex interactions between the immune system and EC, ultimately determining the fate of the tumour. If EC cells manage to create an immunosuppressive microenvironment, they transition into the “escape” phase, evading immune control [
50]. As a result, EC cells regain their ability to proliferate and form distant metastases [
47].
Understanding the mechanism of the shift from the equilibrium to the escape phase holds the potential to inform the development of effective immunotherapies. Recent research suggests that tumour cells release factors such as vascular endothelial growth factor (VEGF), transforming growth factor beta (TGF-β), and indoleamine 2,3-dioxygenase (IDO) to suppress immune cells [
51,
52,
53]. Tumour cells can evade immunosurveillance by shedding tumour antigen and major histocompatibility complex class I molecules or by employing immune-inhibitory mechanisms involving regulatory T cells (Tregs) and myeloid-derived suppressor cells [
54]. Immune checkpoint pathways play a crucial role in inhibiting activated T-cells through negative regulatory pathways, and these pathways must be upregulated by tumour cells to evade immune surveillance [
55]. In EC, the immune system requires two signals to activate naïve T-cells and prompt them to recognise and target tumour cells [
56]. The initial signal involves T-cell receptors (TCR) binding to antigenic peptide-bearing major histocompatibility complexes on EC cells, though this signal alone is insufficient to activate T-cells. The subsequent signal is generated when costimulatory molecules such as CD80 and CD86 (also known as B7-1 and B7-2) on the antigen-presenting cell (APC) interact with T-cell ligands like CD28 [
47,
57,
58]. Immune checkpoint pathways play a role in negatively regulating this two-signal activation process, enabling EC cells to evade immune attack [
59,
60,
61]. Key components of immune checkpoint signalling, such as PD-1, CTLA-4, and PD-L1, are expressed by T cells and other immune cells on the surface of EC cells. CTLA-4 competes with CD28 for B7 ligands, thereby dampening the costimulatory signalling of the CD28/B7 axis (the second signal) [
61,
62,
63]. PD-1/PD-L1 engagement triggers the recruitment of tyrosine phosphatase SHP2, which dephosphorylates proximal signalling regions of T-cell receptors [
64]. This dephosphorylation results in a negative costimulatory effect, suppressing T-cell activation [
61]. These immune checkpoint signals have emerged as crucial targets for novel immunotherapies.
3. Expression of PD-1 and PD-L1 in Endometrial Cancer
PD-1 is a cell surface molecule consisting of 288 amino acids. It is classified as a membrane protein within the immunoglobulin superfamily in humans. PD-1 functions to suppress both adaptive and innate immune responses. This protein is present in various immune cell types, including activated T cells, natural killer (NK) cells, B lymphocytes, macrophages, DCs, and monocytes [
65,
66]. Notably, tumour-specific T lymphocytes exhibit a high expression of PD-1 [
67]. Transcription factors, specifically nuclear factor of activated T cells (NFAT), forkhead box protein (FOX) O1, and interferon (IFN) regulatory factor 9 (IRF9), hold the capacity to initiate the transcription process of PD-1 [
66,
68]. Critical in the regulation of the PD-1 gene’s expression are the upstream regulatory regions B and C, designated as CR-B and COR-C. The CR-C region harbours a binding site associated with NFATc1 (NFAT2) in TCD4 and TCD8 cells [
66,
69]. In contrast, the protein c-FOS engages with specific sites within the CR-B region. This interaction amplifies the production of PD-1 when T-cell receptors are activated upon antigen recognition in naïve T cells. PD-1 exhibits a dualistic nature, encompassing both beneficial and detrimental effects. On the advantageous side, it plays a pivotal role in curbing ineffective or harmful immune responses and upholding immunological tolerance. However, PD-1 activation hampers the protective immune response, contributing to the advancement of malignant cells [
70]. PD-1 is a member of the CD28 family and is encoded by the PDCD1 gene situated on chromosome 2q37.3. This protein has two distinct ligands: PD-L1 and programmed cell death ligand 1 (PD-L2). These ligands exhibit diverse expression patterns and are, respectively, encoded by the CD274 gene and the PDCD1LG2 gene, both located on chromosome 9p24.1 [
71].
PD-L1 is typically detected in macrophages, activated T cells, B cells, DCs, and certain epithelial cells, particularly in the presence of inflammatory stimuli [
66,
72]. Additionally, tumour cells exploit PD-L1 expression as a means to evade anti-tumour responses, representing an adaptive immune mechanism [
73]. The presence of PD-L1 is associated with an immunological microenvironment characterised by an abundance of CD8 T cells, the secretion of Th1 cytokines and chemical factors, and the generation of interferons and distinct gene expression patterns [
74]. Prior studies have provided evidence that interferon-gamma (IFN-γ) triggers an elevation in PD-L1 expression in ovarian cancer cells, contributing to disease progression. Conversely, the inhibition of IFN-γ receptor 1 has been shown to reduce PD-L1 levels in acute myeloid leukaemia mouse models. This reduction is achieved through the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) and myeloid differentiation primary response 88 (MYD88)/Tumor necrosis factor receptor-associated factor 6 (TRAF6) pathways [
11]. The induction of protein kinase D isoform 2 (PKD2) by IFN-γ plays a crucial role in PD-L1 regulation. Inhibiting PKD2 activity suppresses PD-L1 expression, thereby enhancing immune response effectiveness against tumours. Interferon-gamma (IFN-γ) is produced by NK cells through the activation of the Janus kinase (JAK)1, JAK2, and signal transducer and the activator of the transcription (STAT)1 pathway. This leads to an upregulation of PD-L1 expression on the tumour cell surface [
75]. Studies conducted on melanoma cells have demonstrated that T cell-secreted IFN-γ, via the JAK1/JAK2-STAT1/STAT2/STAT3-IRF1 pathway, can modulate PD-L1 expression. Both T cells and NK cells release IFN-γ, leading to the induction of PD-L1 expression on target cell surfaces, including tumour cells [
76].
PD-L1 functions as a promoter of tumour growth within cancer cells by engaging specific receptors and initiating signalling pathways that encourage cell proliferation and survival [
77]. This discovery contributes further evidence to the involvement of PD-L1 in the subsequent progression of tumours. Furthermore, it has been demonstrated that PD-L1 exerts non-immune proliferative effects on various types of tumour cells. For instance, the activation of PD-L1 has been observed to trigger epithelial-to-mesenchymal transition (EMT) and the acquisition of stem cell-like characteristics in renal cancer cells. This implies that the intrinsic pathway of PD-L1 plays a role in facilitating the advancement of kidney cancer [
78].
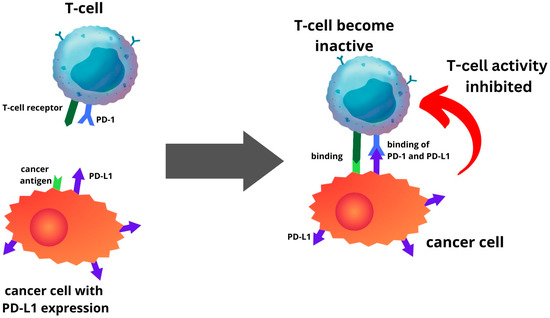
PD-L1 is one of several ligands capable of binding to the PD-1 receptor. In a process where tumour cells elevate PD-L1 production, this protein interacts with PD-1 in T cells, initiating a co-inhibitory signal within these T cells [
13,
79]. This mechanism enables tumour cells to evade elimination through T-cell cytolysis, thereby fostering tumour progression (
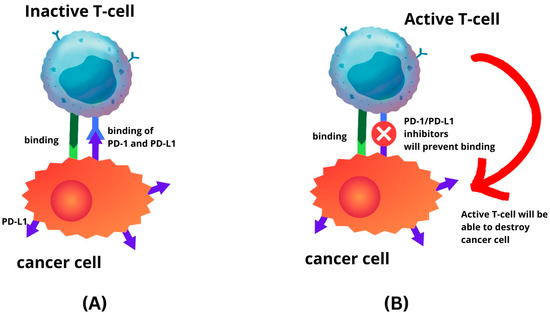
Figure 1). The route is a primary focal point for immune checkpoint inhibitors (ICIs). The PD-1 or PD-L1 inhibitors inhibit the interaction between the PD-L1 and PD-1 receptor. Hence, this prevents cancer cells from escaping the immune system by reactivating the T-cell-mediated process of eliminating tumour cells (
Figure 2). Recent investigations have unveiled that anti-PD-1/PD-L1 first line therapy yields response rates varying between 20% and 65% in PD-L1-positive tumours in various cancers, including EC [
24,
80,
81,
82]. Conversely, tumours lacking PD-L1 expression exhibit response rates ranging from 0% to 17% across diverse tumour types [
79]. The significance of PD-L1 expression within the tumour microenvironment is recognised as a pivotal biomarker for identifying individuals who are more likely to benefit therapeutically from immunotherapy [
13].
Figure 1. Endometrial cancer cells with PD-L1 expression will be able to inhibit T-cell activity by binding the PD-L1 to PD-1 of T-cell.
Figure 2. Immune-checkpoint inhibitor mechanism. (A) Binding of PD-L1 to PD-1 will inhibit T-cell activity; (B) as the result of PD-1 and PD-L1 inhibitors, T-cell will remain active.
The expression levels of PD-1 and PD-L1 in EC are remarkably high, with PD-1 being expressed in approximately 60–65% of EC cases and PD-L1 in a range of 25–70% [
19,
83,
84,
85,
86,
87,
88]. These levels are among the highest reported among gynaecological malignancies. However, ongoing debates persist around the expression patterns of PD-L1 across different molecular subtypes of EC. For instance, in a study by Howitt et al., it was discovered that the frequency of PD-L1 expression was higher in POLE and MSI tumours compared to MSS tumours, particularly in intraepithelial immune cells [
89]. This comparison was made between cases exhibiting the presence versus complete absence of PD-L1 expression.
A study conducted on a cohort of 132 patients diagnosed with MSS, grade 2 endometrioid carcinoma highlighted a specific subgroup within MSS endometrioid carcinomas exhibiting elevated PD-L1 expression [
19]. The results from this study indicated that within the cohort of MSS tumours, approximately 48% of them exhibited positive PD-L1 expression. Moreover, within this group, about 16% expressed a more diffuse or notably intense PD-L1 expression. This subgroup of MSS EC with elevated PD-L1 expression shared an interesting similarity with a distinct class of EC known as MSI EC. These cancers are characterised by a particular microsatellite genetic instability. Remarkably, the subgroup identified in the study not only showed elevated PD-L1 expression, but also demonstrated elevated levels of tumour-associated CD3+ and CD8+ lymphocytes. This heightened immune cell presence is a shared feature with MSI EC. In 2019, the European Society for Medical Oncology (ESMO) conducted a comprehensive analysis examining the correlation between MSI status and PD-L1 expression across various cancer types, including EC [
90]. This analysis provided valuable insights into how these two factors interplay in various cancers and potentially influence treatment strategies. The discoveries from both the initial study on the MSS grade 2 endometrioid carcinoma subgroup and the subsequent broader analysis by ESMO shed light on the intricate relationship between PD-L1 expression, MSI, and immune cell infiltration within different types of cancers.
In EC, only a small fraction, comprising 3.1% of patients, were found to exhibit MSI and a positive PD-L1 status. Among the diverse malignancies assessed, the group of patients with MSI-H combined with a positive PD-L1 status constituted a relatively small proportion. A study by Vanderwalde et al., involving 11,348 cases across 23 different cancer types, found that the prevalence of PD-L1-positive cases was 25.4% in the entire population [
91]. However, within the group of patients with MSI-H, only 26% exhibited PD-L1 positivity. The elevated expression of PD-L1 has been linked to a higher prevalence of patients with high-grade and non-endometrioid EC, as reported in previous studies [
17,
92,
93]. Notably, an association has been established between higher levels of PD-L1 expression in tumour cells and immune cells and the presence of dMMR [
92,
93,
94]. Furthermore, TCGA classification has provided new insights into molecular subtype mutations that altered EC’s immunological profile. Previous studies have demonstrated that PD-L1 expression is significantly more pronounced in the POLE and dMMR subgroups compared to the NSMP and p53 mutation subgroups [
17,
93,
95]. Several studies, including those by Li et al. and Zong et al., have reported associations between PD-L1 expression in immune cells and factors such as deep myometrial invasion, the presence of lymphovascular space invasion (LVSI), and the histological subtype of non-endometrioid EC. However, these associations were not consistently observed in tumour cells [
92,
93]. Additionally, a higher expression of PD-L1 in tumour cells was more frequently observed in high-grade tumours compared to low-grade tumours [
92,
93,
96]. These findings highlighted the complex relationships between microsatellite instability, PD-L1 expression, and various clinicopathological features in EC. Understanding these associations provides valuable insights into the complex molecular landscape that governs the behaviour of different subtypes of EC, as well as prognostic indicators and potential treatment strategies.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242015233