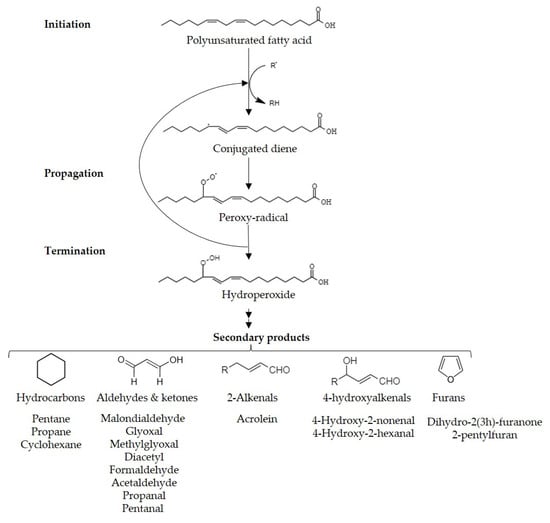
Lipid peroxidation, the most aggressive reaction in food, results in the formation of reactive organic compounds that detrimentally impact food sensory qualities and consumers’ health. While controlled lipid peroxidation can enhance flavors and appearance in certain foods, secondary peroxidation products lead to sensory deterioration in a variety of products, such as oils, alcoholic beverages, and meat. Dispersive liquid-liquid microextraction (DLLME), solid-phase microextraction (SPME), and gas-diffusion microextraction (GDME). These techniques offer efficient and sensitive approaches to extracting and quantifying lipid oxidation products and contribute to the understanding of oxidative deterioration in various food products.
- food analysis
- gas-diffusion microextraction
- lipid peroxidation
- dispersive liquid-liquid microextraction
- solid-phase microextraction
1. Introduction
| Secondary Product | CAS Number | IARC Category | Tolerable Daily Intake µg/Kg bw/Day |
Reference | |
|---|---|---|---|---|---|
| Saturate Carbonyls | Formaldehyde | 50-00-0 | 1 | 150 | [22] |
| Acetaldehyde | 75-07-0 | 2B | 185 a | [23] | |
| Hexanal | 66-25-1 | - | 780 * | [24] | |
| α,β-Unsaturated Carbonyls | Acrolein | 107-02-8 | 2A | 7.5 | [25] |
| 4-hydroxy-2-nonenal | 75899-68-2 | 3 | 1.5 ** | [26] | |
| 4-hydroxy-2-hexenal | 17427-21-3 | 3 | 1.5 ** | [26] | |
| Acrylamide | 79-06-1 | 2A | NE | [27] | |
| Crotonaldehyde | 4170-30-3 | 2B | - | - | |
| Dicarbonyls | Malondialdehyde | 102-52-3 | 3 | 30 ** | [26] |
| Glyoxal | 107-22-2 | - | 200 | [28] | |
| Methylglyoxal | 78-98-8 | 3 | - | - | |
| Diacetyl | 431-03 | - | 900 * | [28] | |
| Furans | Dihydro-2(3H)-furanone | 96-48-0 | 3 | - | - |
| Furfural | 98-01-1 | 3 | 500 | [29] | |
2. Gas Diffusion Microextraction
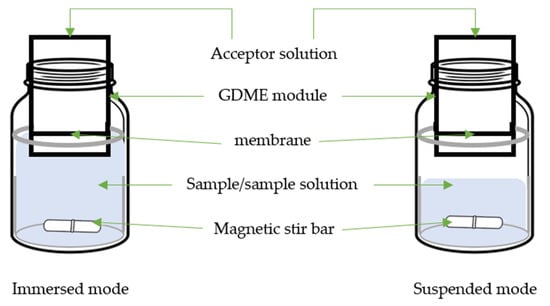
| Target Compound | Sample | GDME | Derivative Reagent | Determination | LOD µg/L or µg/Kg |
Recovery % |
Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Mode | Vacceptor solution mL |
t min |
T °C |
|||||||
| 1,3-pentadione Diacetyl |
Beer | Immersed | 0.5 | 15 | 40 | O-PDA | HPLC-UV | 3.8–4.6 | - | [48] |
| 2 aldehydes & Furfural |
Beer | Immersed | 0.75 | 5 | 30 | DNPH | HPLC-UV | 1.5–12.3 | - | [54] |
| 5 aldehydes | Beer | Suspended | 0.5 | 20 | 40 | HBA | HPLC-DAD | 1.2–1857.7 | >96% | [55] |
| Diacetyl 1 | Wine | Immersed | 0.4 | 20 | 65 | O-PDA | HPLC-UV | 3.8 | - | [56] |
| Acetaldehyde 1 | Wine | Immersed | 1.0 | 15 | 50 | DNPH | HPLC-UV | 800–1100 | - | [57] |
| Diacetyl | Wine & beer | Suspended | 1.0 | 10 | 60 | O-PDA | DPV | 0.053 | - | [58] |
| α-DCC | Wine; black tea & soy sauce | Immersed 2 | 0.5 | 10 | 55 | O-PDA | HPLC-UV | 50–200 | - | [59] |
| MDA | Vegetable oil | Suspended | 0.5 | 30 | 65 | TBA | HPLC-UV/FLD | 250–350 | ≥82% | [60] |
| 4 aldehydes Acrolein & MDA | Vegetable oil | Suspended | 1.0 | 10 | 60 | DPNH | GC-MS | 50–100 | ≥95% | [10] |
| 2 ketones & diacetyl | Ground bread | Suspended | 0.5 | 15 | 65 | O-PDA | HPLC-UV | 6–12 | - | [61] |
| 27 carbonyl compounds 3 | Green & roast coffee beans | Suspended | 0.5 | 16 | 40 | O-PDA | HPLC-DAD | 50–200 | - | [62] |
3. Solid-Phase Microextraction
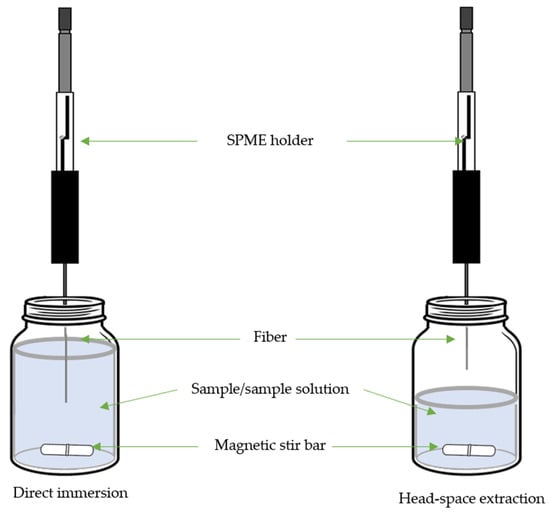
| Target Compound | Sample | SPME | Derivative Reagent | Determination | LOD µg/L or µg/Kg |
Recovery % |
Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mode | t min |
T °C |
Fiber | Tdesorption °C |
|||||||
| 14 aldehydes & ketones | Vegetable oil | HS | 30 | 20 | DVB/CAR/PDMS | 270 | - | GC-FID & GC-MS | 0.04–2.24 | - | [64] |
| 4-HNE | Oils & porcine liver | DI | 15 | 40 | PDMS/DVB | DNPH | HPLC-SP | 0.001–1.42 | 66–87% | [65] | |
| MDA | Cod liver oil | HS | 10 | RT | PDMS/DVB | 200 | N-MH | GC-NPD | 0.74 | 91% | [66] |
| Hexanal | Hazelnut | HS | 10 | 60 | CAR/PDMS | 300 | - | GC-FID | 8.01 | - | [67] |
| 7 aldehydes | Peanut, soybean and olive oils | HS | 15 | 50 | CAR/PDMS | 250 | - | GC-FID | 4.6–10.2 | 85–110 | [68] |
| 3 α,β-UC | Sunflower oil digestion phases | HS | 60 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [69] |
| 100 carbonyl compounds | Cod liver oil | HS | 60 | 50 | DVB/CAR/PDMS | 220 | - | GC-MS | - | - | [70] |
| 18 VOC | Sunflower oil emulsions | HS | 30 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [71] |
| Aldehydes & 2-pentylfuran | Soybean oils | HS | 55 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [72] |
| VOC | Peanut oil | HS | 40 | 50 | PDMS/DVB | 250 | - | GC-MS | - | - | [73] |
| 4 aldehydes & 1 ketone | Roast & boiled duck | HS | 40 | 45 | CAR/PDMS | 280 | - | GC-MS | - | - | [74] |
| 3 aldehydes | Chicken patties | HS | 10 | 60 | DVB/CAR/PDMS | 250 | - | GC-FID | - | - | [75] |
| Hexanal | Pig sausages | HS | 30 | 50 | DVB/CAR/PDMS | 220 | - | GC-MS | - | - | [76] |
| 2 aldehydes & 2 dialdehydes | Cod | HS | 30 | 50 | CAR/PDMS | 260 | - | GC-FID | - | - | [77] |
| 8 aldehydes | Fish | HS | 15 | 60 | PDMS/DVB | 260 | PFBHA | GC-MS | 1.4–6.1 | 79–102 | [78] |
| 6 aldehydes | Caviar | HS | 30 | 60 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [79] |
| 198 VOCs | Dry cured meat | HS | 30 | 37 | 260 | - | GC-MS | - | - | [80] | |
| Aldehydes | Infant formula | HS | 10 | 25 | PDMS/DVB | 250 | - | GC-MS | - | - | [81] |
| 3 aldehydes & pentane | Infant formula | HS | 45 | 37 | CAR/PDMS | 250 | - | GC-FID | 0.02–1.05 | - | [82] |
| 13 Carbonyl compounds | Milk powder | HS | 45 | 43 | 250 | - | GC-MS | 2–6 | - | [83] | |
| VOC | Smoked cheese | HS | 45 | 50 | CAR/PDMS | 260 | - | GC-MS | - | - | [84] |
| VOC | Mozzarella | HS | 15 | 37 | 220 | - | GC-MS | - | - | [85] | |
| VOC | Portuguese cheese | HS | 45 | 50 | DVB/PDMS | 250 | - | GC-MS | - | - | [86] |
| 9 aldehydes | Beer | HS | 60 | 50 | PDMS/DVB | 250 | PFBHA * | GC-MS | - | 89–114 | [87] |
| 41 carbonyl compounds | Beer | HS | 40 | 60 | PDMS/DVB | 250 | PFBHA *,** | GC-MS | 0.003–20,000 | - | [88] |
| 250 carbonyl compounds | Beer | HS | 20 | 45 | PDMS/DVB | 250 | PFBAH ** | GC-ITMS | 0.003–0.510 | 88–114 | [89] |
| 6 carbonyl compound | Beer | HS | 60 | 55 | DVB/CAR/PDMS | 250 | TFEH ** | GC-MS | 0.03–0.5 | 90–105 | [16] |
| 6 carbonyl compound | Craft beer | HS | 60 | 55 | DVB/CAR/PDMS | 250 | TFEH ** | GC-MS | 0.03–0.5 | 90–105 | [90] |
| 18 carbonyl compound | Wine | HS | 45 | 40 | DVB/CAR/PDMS | 250 | - | GC-ITMS | 0.62–129.2 | 19–190 | [91] |
| 80 VOC | Wine | HS | 30 | 40 | DVB/CAR/PDMS | 240 | - | GC-MS | - | - | [92] |
| 6 carbonyl compound | Syrah wines | HS | 45 | 55 | DVB/CAR/PDMS | 250 | TFEH | GCxGC-TOFMS | 0.5–5.2 | 90–106 | [93] |
| 3 aldehydes | Must & wine | HS | 45 | 55 | DVB/CAR/PDMS | 250 | TFEH | GC-qMS | 0.1–0.8 | 90–102 | [94] |
| 38 carbonyl compound | Port wine | HS | 20 | 32 | PDMS/DVB | 250 | PFBHA | GC-MS | 0.006–0.089 | 88–119 | [95] |
| 45 carbonyl compound | Wine | HS | 20 | 40 | PDMS/DVB | 250 | PFBHA | GC-MS/MS | - | 71–146 | [96] |
| 9 aldehydes | Spirits and alcoholic beverages | DI | 15 | 20 | PDMS | 250 | PFBHA | GC-ECD | 0.05–0.5 | - | [97] |
| VOC & SVOC | Beer, wine & whisky | HS | 60 | 30 | PDMS CAR/PDMS DVB/CAR/PDMS |
250 260 260 |
- | GC-MS | - | - | [11] |
| 20 aldehydes | Green pomace distillates | HS | 40 | 55 | PDMS/DVB | 250 | PFBHA | GC-MS | 0.0007–0.02 | 76–110 | [98] |
| 107 VOC | Cider | HS | 30 | 50 | DVB/CAR/PDMS | 250 | - | GC-MS | - | - | [99] |
| 53 carbonyl compounds | Huangjiu (alcoholic beverage) | HS | 35 | 45 | DVB/CAR/PDMS | 250 | PFBHA | GC-MS/MS | - | 71–146 | [100] |
| 2 α-DCC | Soybean paste, red pepper past, soy sauce, wine, beer, distilled liquor | HS | 20 | 85 | DVB/CAR/PDMS | 240 | TFEH | GC-MS | 0.7–1.1 | 92–104 | [101] |
4. Dispersive Liquid-Liquid Microextraction
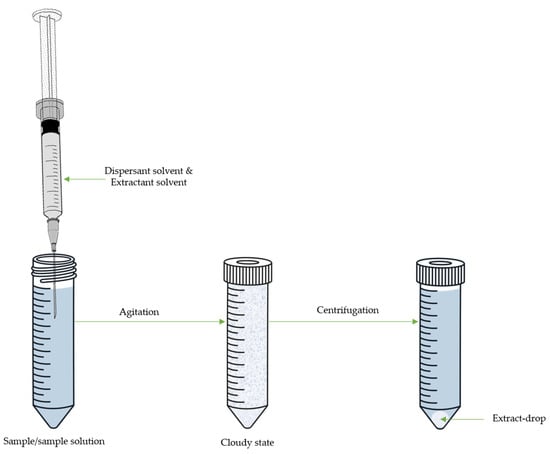
| Target Compound | Sample | DLLME | Derivative Reagent | Determ. | LOD µg/L or µg/Kg |
Rec. % |
Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mode | Disperser | Extracting Solvent | T min |
T °C |
|||||||
| Formaldehyde | Beverages | MW-IL- | ACN | IL 3453W | 1.5 | - | DNPH | HPLC-UV | 0.12 | 85–95 | [112] |
| Acrylamide | Brewed coffee | - | ACN | DCM | - | - | - | UPLC-MS/MS | 900 | 97–106 | [113] |
| PCB and acrylamide | Milk/Coffee | IL | [HeOHMIM][Cl] | [BMIM][NTf2] | - | - | - | HS-GC-ECD-MS | - | - | [114] |
| MDA, acrolein, 4-HNE | Beverages | US | ACN | CH3Cl | 5 | 60 °C | DNPH | GC-MS | 50–200 | 94–102 | [115] |
| Formaldehyde | Milk | IL | MeOH | IL 3453W | 0.75 | 45 °C | ACAC | UV | 100 | 91–103 | [116] |
| Acrylamide | Coffee, chocolate, roasted nuts, French fries, cereals, biscuits, chips, bread, and caramelized fruit |
SSA | SUPRAS-2 (SDS/TBABr/AlCl3) |
2 | - | UV | 0.2 | 93–96 | [117] | ||
| Acrylamide | Nuts and seeds | - | PCE | EtOH | 3 | - | Xanthydrol | GC-MS | 0.6 | 95 | [118] |
| Acrylamide | Potato chips | UAE | PCE | EtOH | 2 | - | Xanthydrol | GC-MS | 0.6 | 97 | [119] |
| Acrylamide | Cereal products | - | PCE | EtOH | 1 | - | Xanthydrol | GC-MS | 0.6 | 95 | [120] |
| Acrylamide | Bread | UAE | PCE | MeOH | 1 | - | Xanthydrol | GC-MS | 0.54 | 98 | [121] |
| 4 aldehydes Acrolein & MDA | Vegetable oil | US | ACN | CH3Cl | 5 | 60 | DPNH | GC-MS | 50–100 | ≥95% | 10 |
5. Combined Microextraction Techniques
This entry is adapted from the peer-reviewed paper 10.3390/separations10100531