Alzheimer’s disease (AD) is the most common cause of dementia, characterised by a marked decline of both memory and cognition, along with pathophysiological hallmarks including amyloid beta peptide (Aβ) accumulation, tau protein hyperphosphorylation, neuronal loss and inflammation in the brain. Additionally, oxidative stress caused by an imbalance between free radicals and antioxidants is considered one of the main risk factors for AD, since it can result in protein, lipid and nucleic acid damage and exacerbate Aβ and tau pathology. Green tea, and its main bioactive compound, epigallocatechin-3-gallate (EGCG), have been targeted as a plausible option for the modulation of AD. Specifically, EGCG acts as an antioxidant by regulating inflammatory processes involved in neurodegeneration such as ferroptosis and microglia-induced cytotoxicity and by inducing signalling pathways related to neuronal survival. Furthermore, it reduces tau hyperphosphorylation and aggregation and promotes the non-amyloidogenic route of APP processing, thus preventing the formation of Aβ and its subsequent accumulation.
- Alzheimer’s disease
- EGCG
- neuroprotection
1. Introduction
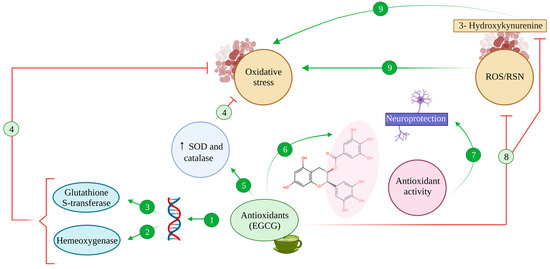
2. Antioxidative Effects of EGCG
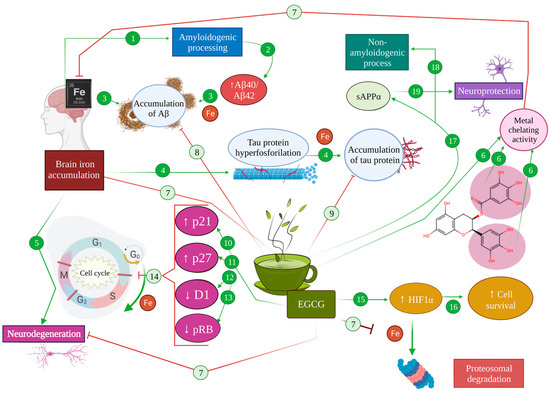
3. Iron-Chelating Effects of EGCG
4. Modulating Effect of EGCG in Cell Signalling, Survival and Death Pathways
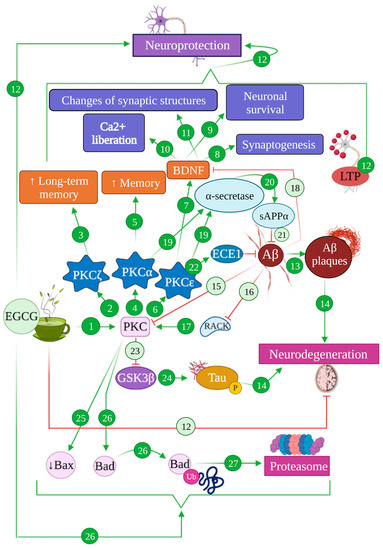
4.1. EGCG and the PKC Pathway: Implications in Alzheimer’s Disease
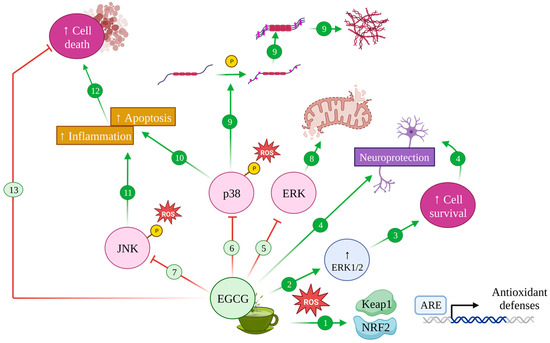
4.2. EGCG and the MAPK Pathway: Implications in Alzheimer’s Disease
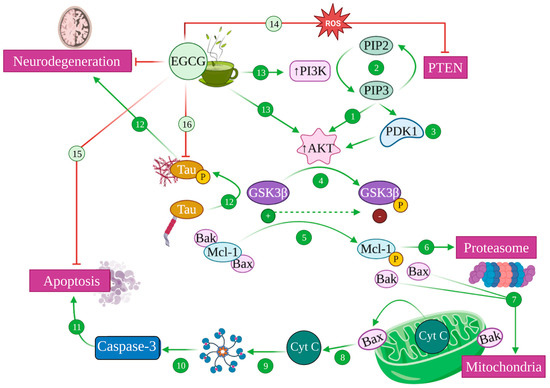
4.3. EGCG and the PI3K/Akt Pathway: Implications in Alzheimer’s Disease
This entry is adapted from the peer-reviewed paper 10.3390/antiox12071460
References
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89.
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2022, 11, 111–128.
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s disease. Neurotherapeutics 2022, 19, 173–185.
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323.
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789.
- Avila, J.; Perry, G. A multilevel view of the development of Alzheimer’s disease. Neuroscience 2021, 457, 283–293.
- Alzheimer’s-Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789.
- Lloret, A.; Esteve, D.; Lloret, M.-A.; Cervera-Ferri, A.; Lopez, B.; Nepomuceno, M.; Monllor, P. When does Alzheimer′ s disease really start? The role of biomarkers. Int. J. Mol. Sci. 2019, 20, 5536.
- Knopman, D.S.; Petersen, R.C.; Jack, C.R. A brief history of “Alzheimer disease”: Multiple meanings separated by a common name. Neurology 2019, 92, 1053–1059.
- Gauthier, S.; Rosa-Neto, P.; Morais, J.; Webster, C. World Alzheimer Report 2021: Journey through the diagnosis of dementia. Alzheimer’s Dis. Int. 2021, 2022, 30.
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau proteins and tauopathies in Alzheimer’s disease. Cell. Mol. Neurobiol. 2018, 38, 965–980.
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Deschaintre, Y.; Richard, F.; Leys, D.; Pasquier, F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 2009, 73, 674–680.
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651.
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105.
- Fernández-Sanz, P.; Ruiz-Gabarre, D.; García-Escudero, V. Modulating effect of diet on Alzheimer’s disease. Diseases 2019, 7, 12.
- Bellantuono, L.; Monaco, A.; Amoroso, N.; Lacalamita, A.; Pantaleo, E.; Tangaro, S.; Bellotti, R. Worldwide impact of lifestyle predictors of dementia prevalence: An eXplainable Artificial Intelligence analysis. Front. Big Data 2022, 5, 1027783.
- Graham, H.; White, P.C.L. Social determinants and lifestyles: Integrating environmental and public health perspectives. Public Health 2016, 141, 270–278.
- Duplantier, S.C.; Gardner, C.D. A critical review of the study of neuroprotective diets to reduce cognitive decline. Nutrients 2021, 13, 2264.
- Burns, S.; Selman, A.; Sehar, U.; Rawat, P.; Reddy, A.P.; Reddy, P.H. Therapeutics of Alzheimer’s Disease: Recent Developments. Antioxidants 2022, 11, 2402.
- Khalatbary, A.R.; Khademi, E. The green tea polyphenolic catechin epigallocatechin gallate and neuroprotection. Nutr. Neurosci. 2020, 23, 281–294.
- Farzaei, M.H.; Bahramsoltani, R.; Abbasabadi, Z.; Braidy, N.; Nabavi, S.M. Role of green tea catechins in prevention of age-related cognitive decline: Pharmacological targets and clinical perspective. J. Cell. Physiol. 2019, 234, 2447–2459.
- Hayat, K.; Iqbal, H.; Malik, U.; Bilal, U.; Mushtaq, S. Tea and its consumption: Benefits and risks. Crit. Rev. Food Sci. Nutr. 2015, 55, 939–954.
- Nabavi, S.M.; Silva, A.S. Nonvitamin and Nonmineral Nutritional Supplements; Academic Press: Cambridge, MA, USA, 2018.
- Urdzikova, L.M.; Ruzicka, J.; Karova, K.; Kloudova, A.; Svobodova, B.; Amin, A.; Dubisova, J.; Schmidt, M.; Kubinova, S.; Jhanwar-Uniyal, M. A green tea polyphenol epigallocatechin-3-gallate enhances neuroregeneration after spinal cord injury by altering levels of inflammatory cytokines. Neuropharmacology 2017, 126, 213–223.
- Mandel, S.A.; Amit, T.; Kalfon, L.; Reznichenko, L.; Youdim, M.B. Targeting multiple neurodegenerative diseases etiologies with multimodal-acting green tea catechins. J. Nutr. 2008, 138, 1578S–1583S.
- Moore, R.J.; Jackson, K.G.; Minihane, A.M. Green tea (Camellia sinensis) catechins and vascular function. Br. J. Nutr. 2009, 102, 1790–1802.
- Lee, M.-J.; Maliakal, P.; Chen, L.; Meng, X.; Bondoc, F.Y.; Prabhu, S.; Lambert, G.; Mohr, S.; Yang, C.S. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: Formation of different metabolites and individual variability. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1025–1032.
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F. Blood brain barrier permeability of (−)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186.
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of green tea catechins in the brain: Epigallocatechin gallate and its metabolites. Int. J. Mol. Sci. 2019, 20, 3630.
- Unno, K.; Pervin, M.; Nakagawa, A.; Iguchi, K.; Hara, A.; Takagaki, A.; Nanjo, F.; Minami, A.; Nakamura, Y. Blood–Brain Barrier Permeability of Green Tea Catechin Metabolites and their Neuritogenic Activity in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2017, 61, 1700294.
- Wyganowska-Świątkowska, M.; Matthews-Kozanecka, M.; Matthews-Brzozowska, T.; Skrzypczak-Jankun, E.; Jankun, J. Can EGCG Alleviate Symptoms Down Syndrome by Altering Proteolytic Act? Int. J. Mol. Sci. 2018, 19, 248.
- Mandel, S.; Weinreb, O.; Amit, T.; Youdim, M.B. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: Implications for neurodegenerative diseases. J. Neurochem. 2004, 88, 1555–1569.
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr. 2009, 4, 283–296.
- Pannala, A.; Rice-Evans, C.A.; Halliwell, B.; Singh, S. Inhibition of peroxynitrite-mediated tyrosine nitration by catechin polyphenols. Biochem. Biophys. Res. Commun. 1997, 232, 164–168.
- Nanjo, F.; Goto, K.; Seto, R.; Suzuki, M.; Sakai, M.; Hara, Y. Scavenging effects of tea catechins and their derivatives on 1, 1-diphenyl-2-picrylhydrazyl radical. Free Radic. Biol. Med. 1996, 21, 895–902.
- Chen, C.; Yu, R.; Owuor, E.D.; Tony Kong, A.-N. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch. Pharmacal Res. 2000, 23, 605–612.
- Owuor, E.D.; Kong, A.-N.T. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770.
- Jeong, J.H.; Kim, H.J.; Lee, T.J.; Kim, M.K.; Park, E.S.; Choi, B.S. Epigallocatechin 3-gallate attenuates neuronal damage induced by 3-hydroxykynurenine. Toxicology 2004, 195, 53–60.
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.; Mandel, S. Green tea polyphenol (–)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem. 2001, 78, 1073–1082.
- Thompson, M.; Williams, C.; Elliot, G. Stability of flavonoid complexes of copper (II) and flavonoid antioxidant activity. Anal. Chim. Acta 1976, 85, 375–381.
- Van Acker, S.A.; Tromp, M.N.; Griffioen, D.H.; Van Bennekom, W.P.; Van Der Vijgh, W.J.; Bast, A. Structural aspects of antioxidant activity of flavonoids. Free Radic. Biol. Med. 1996, 20, 331–342.
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873.
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10.
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520.
- Jellinger, K.; Paulus, W.; Grundke-Iqbal, I.; Riederer, P.; Youdim, M. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J. Neural Transm.-Park. Dis. Dement. Sect. 1990, 2, 327–340.
- Amit, T.; Avramovich-Tirosh, Y.; Youdim, M.B.; Mandel, S. Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J. 2008, 22, 1296–1305.
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 2007, 25, 59–68.
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528.
- Sakaguchi, S.; Furusawa, S.; Yokota, K.; Sasaki, K.i.; Takayanagi, M.; Takayanagi, Y. The Enhancing Effect of Tumour Necrosis Factor-α on Oxidative Stress in Endotoxemia. Pharmacol. Toxicol. 1996, 79, 259–265.
- Mandel, S.A.; Avramovich-Tirosh, Y.; Reznichenko, L.; Zheng, H.; Weinreb, O.; Amit, T.; Youdim, M.B. Multifunctional activities of green tea catechins in neuroprotection. Neurosignals 2005, 14, 46–60.
- Siddiq, A.; Aminova, L.R.; Ratan, R.R. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: Center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem. Res. 2007, 32, 931–946.
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957.
- Ma, Y.; Farny, N.G. Connecting the Dots: Neuronal Senescence, Stress Granules, and Neurodegeneration. Gene 2023, 871, 147437.
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.-g. Cell cycle deregulation in the neurons of Alzheimer’s disease. Cell Cycle Dev. 2011, 53, 565–576.
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668.
- Pastorino, L.; Sun, A.; Lu, P.-J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.-H.; Li, X. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 2006, 440, 528–534.
- Evans, T.A.; Raina, A.K.; Delacourte, A.; Aprelikova, O.; Lee, H.-g.; Zhu, X.; Perry, G.; Smith, M.A. BRCA1 may modulate neuronal cell cycle re-entry in Alzheimer disease. Int. J. Med. Sci. 2007, 4, 140.
- Nurtjahja-Tjendraputra, E.; Fu, D.; Phang, J.M.; Richardson, D.R. Iron chelation regulates cyclin D1 expression via the proteasome: A link to iron deficiency–mediated growth suppression. Blood 2007, 109, 4045–4054.
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505.
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777.
- Wang, J.; Chen, Y.; Chen, L.; Duan, Y.; Kuang, X.; Peng, Z.; Li, C.; Li, Y.; Xiao, Y.; Jin, H. EGCG modulates PKD1 and ferroptosis to promote recovery in ST rats. Transl. Neurosci. 2020, 11, 173–181.
- Plascencia-Villa, G.; Perry, G. Preventive and therapeutic strategies in Alzheimer’s disease: Focus on oxidative stress, redox metals, and ferroptosis. Antioxid. Redox Signal. 2021, 34, 591–610.
- Ma, H.; Dong, Y.; Chu, Y.; Guo, Y.; Li, L. The mechanisms of ferroptosis and its role in Alzheimer’s disease. Front. Mol. Biosci. 2022, 9, 965064.
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938.
- Baptista, F.I.; Henriques, A.G.; Silva, A.M.; Wiltfang, J.; da Cruz e Silva, O.A. Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer’s disease. ACS Chem. Neurosci. 2014, 5, 83–92.
- Lipp, P.; Reither, G. Protein kinase C: The “masters” of calcium and lipid. Cold Spring Harb. Perspect. Biol. 2011, 3, a004556.
- Alkon, D.L.; Sun, M.-K.; Nelson, T.J. PKC signaling deficits: A mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007, 28, 51–60.
- Sun, M.-K.; Alkon, D.L. The “memory kinases”: Roles of PKC isoforms in signal processing and memory formation. Prog. Mol. Biol. Transl. Sci. 2014, 122, 31–59.
- Sun, M.-K.; Alkon, D.L. Dual effects of bryostatin-1 on spatial memory and depression. Eur. J. Pharmacol. 2005, 512, 43–51.
- Hongpaisan, J.; Alkon, D.L. A structural basis for enhancement of long-term associative memory in single dendritic spines regulated by PKC. Proc. Natl. Acad. Sci. USA 2007, 104, 19571–19576.
- Hongpaisan, J.; Sun, M.-K.; Alkon, D.L. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643.
- Sun, M.-K.; Hongpaisan, J.; Alkon, D.L. Postischemic PKC activation rescues retrograde and anterograde long-term memory. Proc. Natl. Acad. Sci. USA 2009, 106, 14676–14680.
- Pakaski, M.; Balaspiri, L.; Checler, F.; Kasa, P. Human amyloid-β causes changes in the levels of endothelial protein kinase C and its α isoform in vitro. Neurochem. Int. 2002, 41, 409–414.
- Desdouits, F.; Buxbaum, J.D.; Desdouits-Magnen, J.; Nairn, A.C.; Greengard, P. Amyloid β peptide formation in cell-free preparations: Regulation by protein kinase C, calmodulin, and calcineurin. J. Biol. Chem. 1996, 271, 24670–24674.
- Hama, H.; Hara, C.; Yamaguchi, K.; Miyawaki, A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 2004, 41, 405–415.
- Stevens, C.F.; Sullivan, J.M. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 1998, 21, 885–893.
- Okada, M.; Zhu, G.; Yoshida, S.; Hirose, S.; Kaneko, S. Protein kinase associated with gating and closing transmission mechanisms in temporoammonic pathway. Neuropharmacology 2004, 47, 485–504.
- Dobransky, T.; Doherty-Kirby, A.; Kim, A.-R.; Brewer, D.; Lajoie, G.; Rylett, R.J. Protein kinase C isoforms differentially phosphorylate human choline acetyltransferase regulating its catalytic activity. J. Biol. Chem. 2004, 279, 52059–52068.
- Meiri, N.; Sun, M.-K.; Segal, Z.; Alkon, D.L. Memory and long-term potentiation (LTP) dissociated: Normal spatial memory despite CA1 LTP elimination with Kv1. 4 antisense. Proc. Natl. Acad. Sci. USA 1998, 95, 15037–15042.
- Sacktor, T.C. Memory maintenance by PKMζ—An evolutionary perspective. Mol. Brain 2012, 5, 31.
- Shema, R.; Haramati, S.; Ron, S.; Hazvi, S.; Chen, A.; Sacktor, T.C.; Dudai, Y. Enhancement of consolidated long-term memory by overexpression of protein kinase Mζ in the neocortex. Science 2011, 331, 1207–1210.
- Basu, A.; Pal, D. Two faces of protein kinase Cδ: The contrasting roles of PKCδ in cell survival and cell death. Sci. J. 2010, 10, 2272–2284.
- Khadra, N.; Bresson-Bepoldin, L.; Penna, A.; Chaigne-Delalande, B.; Ségui, B.; Levade, T.; Vacher, A.-M.; Reiffers, J.; Ducret, T.; Moreau, J.-F. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) β2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19072–19077.
- Adasme, T.; Haeger, P.; Paula-Lima, A.C.; Espinoza, I.; Casas-Alarcón, M.M.; Carrasco, M.A.; Hidalgo, C. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc. Natl. Acad. Sci. USA 2011, 108, 3029–3034.
- Sun, M.-K.; Alkon, D.L. Activation of protein kinase C isozymes for the treatment of dementias. Adv. Pharmacol. 2012, 64, 273–302.
- Choi, D.-S.; Wang, D.; Yu, G.-Q.; Zhu, G.; Kharazia, V.N.; Paredes, J.P.; Chang, W.S.; Deitchman, J.K.; Mucke, L.; Messing, R.O. PKCε increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8215–8220.
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33, S141–S144.
- Eckman, E.; Eckman, C. Aβ-degrading enzymes: Modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1101–1105.
- Kim, T.; Hinton, D.J.; Choi, D.-S. Protein kinase C-regulated Aβ production and clearance. Int. J. Alzheimer’s Dis. 2011, 2011, 857368.
- Eckman, E.A.; Watson, M.; Marlow, L.; Sambamurti, K.; Eckman, C.B. Alzheimer’s disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. 2003, 278, 2081–2084.
- Zhu, G.; Wang, D.; Lin, Y.-H.; McMahon, T.; Koo, E.H.; Messing, R.O. Protein kinase C ϵ suppresses Aβ production and promotes activation of α-secretase. Biochem. Biophys. Res. Commun. 2001, 285, 997–1006.
- Funalot, B.; Ouimet, T.; Claperon, A.; Fallet, C.; Delacourte, A.; Epelbaum, J.; Subkowski, T.; Leonard, N.; Codron, V.; David, J. Endothelin-converting enzyme-1 is expressed in human cerebral cortex and protects against Alzheimer’s disease. Mol. Psychiatry 2004, 9, 1122–1128.
- Behl, C.; Ziegler, C. Beyond amyloid–widening the view on Alzheimer’s disease. J. Neurochem. 2017, 143, 394–395.
- Zhang, T.; Chen, D.; Lee, T.H. Phosphorylation signaling in APP processing in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 21, 209.
- Kalfon, L.; Youdim, M.B.; Mandel, S.A. Green tea polyphenol (–)-epigallocatechin-3-gallate promotes the rapid protein kinase C-and proteasome-mediated degradation of Bad: Implications for neuroprotection. J. Neurochem. 2007, 100, 992–1002.
- Rezai-Zadeh, K.; Shytle, D.; Sun, N.; Mori, T.; Hou, H.; Jeanniton, D.; Ehrhart, J.; Townsend, K.; Zeng, J.; Morgan, D. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 2005, 25, 8807–8814.
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882.
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 463–475.
- Takeda, K.; Naguro, I.; Nishitoh, H.; Matsuzawa, A.; Ichijo, H. Apoptosis signaling kinases: From stress response to health outcomes. Antioxid. Redox Signal. 2011, 15, 719–761.
- Newlaczyl, A.U.; Hood, F.E.; Coulson, J.M.; Prior, I.A. Decoding RAS isoform and codon-specific signalling. Biochem. Soc. Trans. 2014, 42, 742–746.
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245.
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692.
- Mansuri, M.L.; Parihar, P.; Solanki, I.; Parihar, M.S. Flavonoids in modulation of cell survival signalling pathways. Genes Nutr. 2014, 9, 400.
- Chlan-Fourney, J.; Zhao, T.; Walz, W.; Mousseau, D. The increased density of p38 mitogen-activated protein kinase-immunoreactive microglia in the sensorimotor cortex of aged TgCRND8 mice is associated predominantly with smaller dense-core amyloid plaques. Eur. J. Neurosci. 2011, 33, 1433–1444.
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.-W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706.
- Gruendler, R.; Hippe, B.; Sendula Jengic, V.; Peterlin, B.; Haslberger, A.G. Nutraceutical approaches of autophagy and neuroinflammation in Alzheimer’s disease: A systematic review. Molecules 2020, 25, 6018.
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877.
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549.
- Gan, X.; Huang, S.; Wu, L.; Wang, Y.; Hu, G.; Li, G.; Zhang, H.; Yu, H.; Swerdlow, R.H.; Chen, J.X. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 220–231.
- Na, H.-K.; Kim, E.-H.; Jung, J.-H.; Lee, H.-H.; Hyun, J.-W.; Surh, Y.-J. (−)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch. Biochem. Biophys. 2008, 476, 171–177.
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116.
- Choi, Y.-J.; Jeong, Y.-J.; Lee, Y.-J.; Kwon, H.-M.; Kang, Y.-H. (-) Epigallocatechin gallate and quercetin enhance survival signaling in response to oxidant-induced human endothelial apoptosis. J. Nutr. 2005, 135, 707–713.
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 2019, 35, 775–795.
- Brazil, D.P.; Yang, Z.-Z.; Hemmings, B.A. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 2004, 29, 233–242.
- Bayascas, J.R.; Alessi, D.R. Regulation of Akt/PKB Ser473 phosphorylation. Mol. Cell 2005, 18, 143–145.
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100, 603–606.
- Eldeeb, M.A.; Esmaili, M.; Hassan, M.; Ragheb, M.A. The role of PTEN-L in modulating PINK1-Parkin-mediated mitophagy. Neurotox. Res. 2022, 40, 1103–1114.
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71.
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131.
- Sayas, C.L.; Ávila, J. GSK-3 and tau: A key duet in alzheimer’s disease. Cells 2021, 10, 721.
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 35.
- Koh, S.-H.; Kim, S.H.; Kwon, H.; Park, Y.; Kim, K.S.; Song, C.W.; Kim, J.; Kim, M.-H.; Yu, H.-J.; Henkel, J.S. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Mol. Brain Res. 2003, 118, 72–81.