Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Genetics & Heredity
Large musculoaponeurotic fibrosarcoma (MAF) transcription factors contain acidic, basic, and leucine zipper regions. Four types of MAF have been elucidated in mice and humans, namely c-MAF, MAFA, MAFB, and NRL.
- large MAF transcription factors
- c-MAF
- MAFA
- MAFB
1. Introduction
Medical technology has rapidly advanced in recent years, leading to the identification of the causative genes for many conditions, including cardiovascular disease, diabetes, and malignancies, as well as more than 7000 rare diseases. Despite the limited number of patients with rare diseases and challenges in identifying effective treatment strategies, rapid advances in genome editing tools have accelerated the knowledge of human disease studies owing to our ability to generate diverse genetically modified animal models. Given the advances in gene editing tools, these animal models are gradually progressing toward mimicking human diseases by introducing human pathogenic mutations into their genomes, along with conventional gene knockouts.
The roles of large musculoaponeurotic fibrosarcoma (MAF) transcription factors, including c-MAF, MAFA, and MAFB, in various organs and tissues have been elucidated in both knockout and conditional knockout mice. In addition, point mutations in these genes have recently been reported to cause various rare diseases, such as congenital cataracts, Aymé–Gripp syndrome, multicentric carpotarsal osteolysis (MCTO), Duane syndrome, and focal segmental glomerulosclerosis (FSGS). Therefore, considering that large MAF transcription factors are highly conserved between humans and mice, mouse models that incorporate human pathogenic mutations into these genomes are crucial to understanding the pathophysiological mechanisms and therapeutic strategies for rare diseases.
2. Large MAF Transcription Factors
MAF transcription factors are detected as cellular homologs of v-MAF, an oncogene that was discovered in the avian transforming retrovirus, AS42. The virus causes musculoaponeurotic fibrosarcoma in chickens [1]. The transcription factors, which are called large Maf proteins, consist of a family of transcription factors characterized by a transcriptional activation domain, a basic region required for DNA binding, and a typical bZip structure (bZip domain) which is a motif for protein dimerization and DNA binding (Figure 1A). There are four types of large MAF transcription factors, namely c-MAF, MAFA, MAFB, and NRL; in addition, they have been reported in mice and humans [2][3]. These four transcription factors are thought to be involved in the formation of homodimers and binding to DNA sequences called Maf recognition elements (MAREs) to stimulate the transcriptional activity of nearby target genes. Although the MARE sequence is 13 or 14 bp, it has been reported that half of the sequence (half MARE) activates target genes (Figure 1B) [4]. MAF transcription factors are involved in the important functions of several distinct developmental processes, establishment of the function of cells, tissues, and organs, cell differentiation, and maintaining the function of cells and organs. In recent years, various diseases have been reported in human patients with point mutations in c-MAF, MAFA, and MAFB.
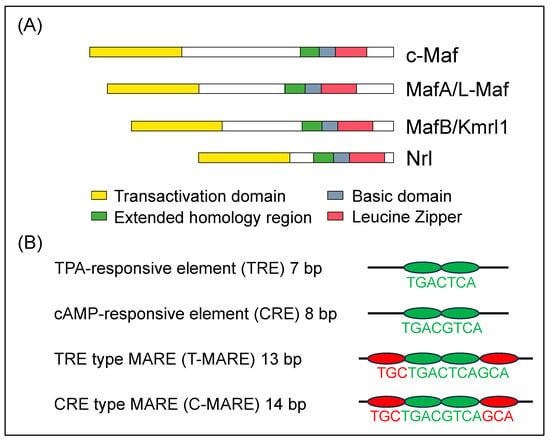
Figure 1. (A) Diagrammatic representation of the molecular structure of human large MAF transcription factors, c-MAF, MAFA, MAFB, and NRL. The structural domains are the transactivation domain (in yellow), extended homology region (in green) and basic region (in blue) in the DNA-binding domain, and leucine zipper (in red) and are shown in boxes [5]. (B) Consensus sequences recognized by bZip transcription factors. There are two types of responsive elements such as TRE and CRE which are the core sequence of the MARE (in green). They are included within T-MARE and C-MARE, respectively. Three bases on the side of TRE and CRE are the flanking sequence (in red), which plays a critical role in the recognition of the MARE [6].
3. Roles of c-MAF
c-MAF is the cellular homolog of v-MAF. It plays an essential role in the IL-4 expression in helper T cells (Th) of the immune system [7]. Several previous studies have demonstrated the significance of c-MAF in the regulation of the functions of various immune cells, particularly T cells. For example, the scaffold protein CARMA1 and IKKβ, two essential regulators of the transcription factor nuclear factor κB (NF-κB), activate c-MAF expression. Through the stimulation of the T-cell receptor, increased c-MAF expression results in the production of cytokines [8]. The loss of c-MAF induces a defect in the number of Th-17 and T cells, suggesting that c-MAF controls the expansion of both Th-17 and T cells [9]. Subsequently, transcriptome analysis has shown that c-MAF plays an essential role in the differentiation of Th17 [10]. In the case of macrophages, c-MAF controls the expression of IL-10, which is involved in the differentiation of regulatory T cells [11]. In addition to immune cells, abnormal structures of the eye lens have also been identified in c-Maf homozygous-deficient mice. For example, a detailed analysis of the lens in c-Maf homozygous knockout mice confirmed that there was no expression of CRYSTALLIN, which is a structural protein found in the lens, due to the presence of the MARE sequence in Crystallin genes. These results suggest that c-MAF plays an important role in lens formation by directly regulating the gene expression of Crystallin [12][13]. Furthermore, c-Maf is regulated by p53 together with Prox-1, and as a result, the expression of various Crystallin genes is regulated [14]. In the case of cell differentiation, c-MAF plays an essential role in the differentiation of lens fiber cells to lens epithelial cells, resulting in epithelial cells spreading in the anterior and posterior lens [15]. Other MAFs, such as MAFA and MAFB, are not required for lens fiber cell differentiation [16]. Furthermore, fetal analysis identified the important role of c-MAF in bone formation [17].
The roles of c-MAF in the immune system and developmental stages have mainly been discovered through studies on c-Maf homo-deficient mice. These mice are embryonic lethal due to an abnormal formation of blood islands in the liver in the embryo [18]. To address this issue, conditional knockout mice using the CRISPR-Cas9 genome editing tool were generated, and their functions in the adult stage were clarified. When adult c-MAF conditional knockout mice were systemically deficient in c-MAF, they survived; however, they developed cataracts owing to lens maintenance failure. Detailed lens analysis revealed that the abnormal lens structure and onset of cataracts were caused by the inability to differentiate from lens epithelial cells into lens fiber cells. To researchers' knowledge, this is the first study to demonstrate the role of c-MAF in adults [19]. Subsequently, other studies have demonstrated inhibition of the development of the liver sinusoid by specific deletion of c-MAF in endothelial cells, which induces aberrant expansion of postnatal liver hematopoiesis, accelerates excessive proliferation of sinusoidal cells during the postnatal period, and worsens sensitivity of pro-fibrotic effects in the liver to chemical insults [20]. Moreover, the mechanistic roles of c-MAF in the recovery from acute injury in the intestine promote the differentiation of epithelial cells and the importance of crosstalk between differentiated tuft cells and enterocytes to accelerate long-term intestinal accommodation [21]. While these two studies suggested a requirement for c-MAF in organ injury, we report a protective effect of c-MAF deficiency against diabetes and diabetes-induced nephrophthy through c-MAF regulation of the glucose transporters SGLT2 and GLUT2, which are expressed in the proximal tubules of kidneys[22]. In summary, c-MAF deficiency is interesting and has the potential to decrease susceptibility and provide protection against injury and diseases.
4. c-MAF Point Mutation in Human Patients
Conditions caused by a point mutation in c-MAF were first discovered in 2002, wherein patients showed congenital cataracts and iris hypoplasia [23]. In 2006, patients with pulverulent cataracts, cataracts with microcornea, iris coloboma, and congenital cataracts were reported [24][25]. Moreover, the vibratory sensation was reduced in patients with the same arginine-to-proline mutation in the DNA-binding region of c-MAF as in human cases discovered in 2002 [26]. All these cases were caused by the inability of c-MAF to bind to the MARE sequences of the target genes due to mutations in the DNA-binding site of c-MAF, in which arginine was changed into proline (Figure 2).
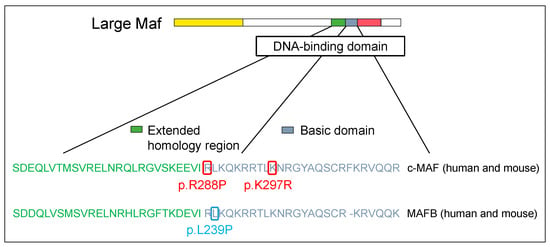
Figure 2. Large Maf DNA-binding domain mutations in human disease. The structural domains are the transactivation domain (in yellow), extended homology region (in green) and basic region (in blue) in the DNA-binding domain, and leucine zipper (in red).
For mutations in the transcriptional activation domain, c-MAF was detected as the causative gene for Aymé–Gripp syndrome resulting in congenital cataracts, deafness, mental retardation, epilepsy, and skeletal dysplasia [27]. Since then, various conditions such as dental abnormalities, calcifications of the bilateral putamen nucleus, pericardial effusion, femoral neck fracture, and exostosis of the distal phalanx have been reported [28][29][30][31]. Here, a case of Aymé–Gripp syndrome was reported to have a mutation in the phosphorylation residues of the transcriptional activation region, such as Ser54, Thr58, Pro59, Ser62, and Pro69 (Figure 3). Interestingly, this abnormal phosphorylation has been reported in cases of human disease mutations of MAFA and MAFB, both in heterozygous and homozygous individuals. Although only one MAFA mutation has been identified, several MAFB mutations have been identified in the transcriptional activation domain, including p.Ser54Trp, p.Pro59Leu, p.Thr62Ile/Pro, p.Pro63Leu/Arg, p.Ser65Ile, p.Ser66Cys, p.Ser69Leu, p.Ser70Leu/Ala, and p.Pro71Ser. These phosphorylation residues are preserved in human MAF transcription factors, c-MAF, MAFA, and MAFB, and are strongly associated with the development of human diseases.
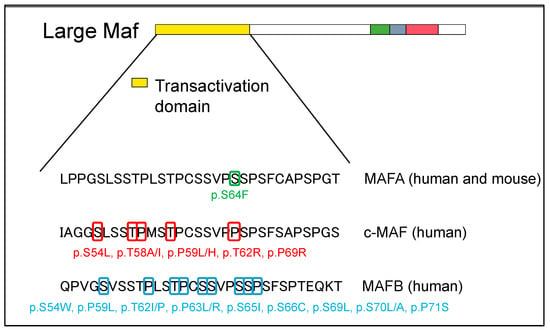
Figure 3. Large MAF phosphorylation site mutations on transactivation domain in human disease. The structural domains are the transactivation domain (in yellow), extended homology region (in green) and basic region (in blue) in the DNA-binding domain, and leucine zipper (in red).
5. Model Mouse of c-MAF Human Point Mutation
To the best of our knowledge, we successfully generated a c-MAF point-mutation mouse for the first time[32]. To investigate the underlying mechanisms of disease onset and identify previously unknown diseases in human patients, a mouse model with a mutation in c-MAF on the GSK3 phosphorylation site, i.e., p.Thr58Ile was generated using the CRISPR-Cas9 system.
The results of sequencing, PCR analysis, IHC staining, and the increased levels and expansion of c-MAF in c-Maf heterozygous mice were compared to those for c-Maf wild-type mice. Moreover, to quantify both RNA and protein expression levels of c-Maf in the kidneys, quantitative PCR (qPCR) analysis and Western blotting were performed. These results showed no significant difference between c-Maf heterozygous and wild-type mice in RNA levels. These results suggest that the increased unphosphorylated protein expression of c-MAF in c-Maf heterozygous mice, compared to that in wild-type mice, may be attributed to impaired phosphorylation of c-MAF and declined degradation of the c-MAF proteins with the mutation. Therefore, these results confirm the successful insertion of the c-Maf mutated allele into the c-Maf target region, resulting in elevated expression of c-MAF in c-Maf heterozygous mice, compared to that in wild-type mice.
The experimental model mouse exhibited phenotypes similar to those observed in human cases, including lens abnormalities, growth retardation, abnormal skull morphology, and short stature. It should be noted that a phenotype was found that was not observed previously in human patients. First, the c-Maf homozygous mutation mouse embryos became embryonic lethal, similar to the c-Maf homozygous knockout embryos. The c-Maf homozygous mutations induce changes in appearance such as shorter limb sizes and growth retardation of the body in the embryonic stage. This underpins the reason why homozygous patients have not yet been identified. Another difference was in the brain weight between c-MAF-mutant mice and control mice. Although patients with c-MAF mutations exhibit intellectual and developmental disabilities, the reason for this has not yet been elucidated. Therefore, this mouse model may be useful for future investigations to assess whether this difference is due to a smaller skull size, a decrease in brain weight, or other underlying factors that are currently unclear. Although Aymé–Gripp syndrome is rare, the experimental murine model may be used to discover previously unreported syndromes in patients and serve as an ideal model for establishing new treatments. Therefore, these kinds of point mutation mouse models are valuable because they can provide novel functional insights into both humans and mice and can be used to analyze the functional roles of target genes.
6. Roles of MAFA
MAFA is detected as a transcription factor inducing the development of the lens of eyes in the epidermis of chickens [33]. Further studies showed that MAFA activates the insulin gene C1 element through the binding of the promoter site, contributing to the increased expression, function, and differentiation of insulin in β cells of the pancreas [34][35]. MAFA coordinates with MAFB, a transactivator of cells, acting on the glucagon gene G1 element, and in conjunction with other transcription factors and related genes to induce the generation and differentiation of β cells [36][37]. In subsequent studies, MAFB was identified in both α and β cells during the early developmental stage. Following a reduction in MAFB expression, MAFA is mainly expressed instead of MAFB [38][39][40]. However, while Mafa homo-deficient mice were born normally, in contrast to c-MAF-knockout mice, they subsequently developed mild fasting hyperglycemia. Further, the mice exhibited abnormal glucose, and histological analysis of the islets in the pancreas revealed abnormal strictures in the adult stage. Gene expression analysis identified various genes, such as Ins1, Ins2, Pdx1, Glut2, and Glanuphilin, as target genes of MAFA [41][42]. Although the expression profile of MAFA indicates that it is highly expressed in the cerebellum, skeletal muscle, thyroid, and testes, the detailed roles of MAFA remain unclear, except in the lens and pancreatic islets.
7. MAFA Point Mutation in Human Patients
In 2018, two unrelated families with point mutations in the transcriptional activation domain of the MAFA gene were reported to have familial diabetes and insulinoma. In addition, the report showed that insulinomatosis was more frequent in females and diabetes was more frequent in males. A missense mutation of MAFA (p.Ser64Phe, c.191C > T) induces the phenotypes of both insulinomatosis and diabetes; in addition, the mutation was found to decrease phosphorylation within the transactivation region of MAFA, similar to a mutation of c-MAF on a GSK3 phosphorylation site (Figure 3). Further, the protein stability of MAFA profoundly increased under both high and low concentrations of glucose in β-cell lines. Therefore, phenotypes in human cases with the missense mutation of MAFA reflect both the oncogenic capacity of MAFA and its important role in the activity of pancreatic β-cells [43].
8. Model Mouse of MafA Human Point Mutation
In 2021, since the number of human patients with MAFA mutation is small and mechanistic studies in humans are difficult, CRISPR-Cas9 gene editing was used to generate an experimental mouse model harboring the same pathogenic change of a single base pair as human patients [44]. This heterozygous mutant male showed impaired glucose tolerance, whereas heterozygous mutant female mice showed slight hypoglycemia. Surprisingly, protein expression of MAFA in four-week-old heterozygous mutant male mice was significantly increased one week before the onset of glucose intolerance. In contrast, heterozygous mutant female mice did not show elevated expression. This finding may explain the different phenotypes observed in heterozygous mutant male and female mice.
Notably, differences exist between c-MAF- and MAFB-mutant mice and MAFA-mutant mice in terms of their proclivity to develop insulinoma, a typical disease in humans. Although female mutant mice displayed slight hypoglycemia and the amelioration of glucose tolerance, the previously mentioned study did not confirm whether insulinoma development occurred or the reasons for it. Further, c-MAF- and MAFB-mutant mice exhibited similar phenotypes to human patients, suggesting the need to create conditions conducive to insulinoma development. Nevertheless, these mice are useful experimental models for diabetes research due to their abnormal β cells.
9. Roles of MAFB
The Mafb gene was detected in Kreisler (Kr) mice, showing turning behavior resulting from impairment of the development of the inner ear by radiation. Further study indicated that MafB plays an important role in segment formation in the hindbrain; in addition, the absence of MafB induces abnormal formation of the inner ear [45]. MAFB was isolated through homology screening for v-MAFs [46] and was found to inhibit the function of ETS1 and the differentiation of erythroid cells in the hematopoietic system in chickens [47]. Moreover, overexpression of MAFB stimulates the differentiation of both macrophages and monocytes [48]. Notably, a previous study showed that krml homozygous mutant mice develop nephrotic syndrome resulting from glomerular podocyte impairment. In addition, the study highlighted that MAFB was shown to be essential for the differentiation of the epithelial cells of the podocytes during the final stages of development [49].
Similar to the previous study, Mafb-Gfp knock-in/knock-out mice died immediately after birth due to hypoplasia of the hindbrain, and kidney and respiratory failure. However, the number of macrophages expected to increase, as in the previous study [48], was normal. Mafb knockout suppressed F4/80 expression in mature macrophages, resulting in renal dysgenesis with abnormal podocyte differentiation and tubular apoptosis [50]. Subsequently, it was reported that although there was no abnormality in the differentiation of macrophages, there was an abnormality in the formation of actin in macrophages following the deletion of MAFB [51]. Furthermore, Mafb heterozygous mice showed abnormal parathyroid gland formation, while homozygous-deficient mice did not produce parathyroid hormone [52]. These results suggest diverse roles of MAFB in mice. In addition, it was reported that MAFB is involved in various biological processes, including the myeloid commitment of hematopoietic stem cells, growth of macrophages [53][54], generation of the thymus [55], creation of hair cuticles [56], differentiation of skin keratinocytes [57], formation of the urethra [58], and lymphangiogenesis [59].
MAFB regulates the phagocytic function of macrophages by regulating the expression of C1q, which is a complement component essential for the phagocytosis of foreign substances and dead cells, and the expression of MSR1, which is a scavenger receptor [60][61]. MAFB is necessary for the creation and maintenance of podocytes in kidneys at the embryonic [49] and adult stages, respectively [62]. In 2007, it was reported that MAFB is expressed in α and β cells of the mouse fetal pancreas and Mafb homozygous-deficient mice show suppressed development of α and β cells in the pancreas during the embryonic stage [39]. Although MAFA is essential for the expression of insulin in β cells of the pancreas, using conditional knockout mice, it was revealed that MAFB also contributes to pancreatic β-cell development during the embryonic phase. While MAFB is inactive under normal conditions, it functions under pathological conditions, underpinning its importance in the development and maintenance of cell functions [63][64].
10. MAFB Point Mutation in Human Patients
Since c-MAF plays important roles in lens, bone, liver, and kidney formation, and patients with c-MAF mutations show many disorders, such as congenital cataracts, deafness, skeletal dysplasia, dental abnormalities, epilepsy, mental retardation, calcifications of the bilateral putamen nucleus, pericardial effusion, exostosis of the distal phalanx, and femoral neck fracture [21][23][24][25][65], it was considered that MAFB mutations also cause various diseases. Since Mafb homozygous knockout mice die immediately after birth due to respiratory failure, it was thought that Mafb heterozygous mutant patients might have abnormalities in their respiratory organs, followed by several other organs, such as the ears, parathyroid glands, and kidneys, as demonstrated in MAFB-knockout and conditional knockout mice.
In 2012, the results of whole exome sequencing of human cases identified MAFB as the causative gene for multicentric carpotarsal osteolysis (MCTO), which is a syndrome showing the dissolution of the bones of the palm and foot [66]. Similar to c-MAF and MAFA mutation cases, MCTO human patients harbor mutations at the phosphorylation site of the transcriptional activation region, resulting in c.176C > T (p.Pro59Leu), and abnormal phosphorylation inhibiting the degradation of MAFB (Figure 3). In addition to abnormal bone formation, MCTO cases show hereditary FSGS.
Following this discovery, not only patients with mutations in the activation domain but also those with mutations in the binding region were found to be similar to c-MAF. One previous study in 2016 detected MAFB as the causative gene of Duane syndrome, which is characterized by abnormal eye abduction. In Duane syndrome, the cleaved MAFB protein may be encoded by the mutated MAFB gene [67]. The mutation is a highly preserved leucine-to-proline substitution in the DNA-binding region of MAFB, rendering it incapable of binding to the MARE sequence. Finally, podocytes from neonatal mice with the p.Leu239Pro mutation exhibited impaired differentiation. Therefore, mutations of MAFB impair the generation and/or inhibit the sustenance of abducens neurons, the inner ear, and podocytes in the kidneys [68].
11. Model Mouse of MAFB Human Point Mutation
A mouse model with MCTO was generated using CRISPR-Cas9 gene editing [69]. Mafb homozygous mutant mice exhibited MCTO and nephropathies such as glomerular sclerosis and renal failure, developmental defects in body weight, and high levels of urine albumin and creatinine, which are similar to the symptoms found in MCTO human cases. In the clinical setting, variability among patients in diagnosis has been reported; however, this has occurred for unknown reasons [70]. Thus, it is necessary to establish other MCTO mouse models with various mutations to evaluate target genes surrounding MAFB and clarify the implications of these reasons. In MCTO patients, glomerular sclerosis and renal failure frequently occur, and MCTO is diagnosed in the later stages of renal failure. Investigating its indications at earlier stages in Mafb homozygous mutant mice with MCTO can aid in developing diagnostic biomarkers to prevent the progression of nephropathy in patients with MCTO. To identify the pathogenicity of the heterozygous substitution of p.Leu239Pro, an experimental model mouse carrying the MAFB p.Leu239Pro mutation was established using the CRISPR-Cas9 system [68]. Mafb homozygous mutant mice died within 48 h after birth, and Mafb homozygous newborn mice showed defective differentiation of podocytes resulting from the impairment of foot processes. In contrast, Mafb heterozygous mutant mice could stay healthy without any abnormal podocyte formation or kidney function.
Current methods used to treat MCTO nephropathy are invasive and cause severe side effects, with limited application. The usefulness of the two mouse models enabled us to develop alternative treatments, especially for MCTO nephropathy. Moreover, MCTO pathogenesis can be elucidated using an MAFB-mutant mouse model, which may contribute to the generation of novel treatments for MCTO and nephropathy.
This entry is adapted from the peer-reviewed paper 10.3390/genes14101883
References
- Nishizawa, M.; Kataoka, K.; Goto, N.; Fujiwara, K.T.; Kawai, S. v-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc. Natl. Acad. Sci. USA 1989, 86, 7711–7715.
- Motohashi, H.; Shavit, J.A.; Igarashi, K.; Yamamoto, M.; Engel, J.D. The world according to maf. Nucleic Acids Res. 1997, 25, 2953–2959.
- Blank, V.; Andrews, N.C. The maf transcription factors: Regulators of differentiation. Trends Biochem. Sci. 1997, 22, 437–441.
- Motohashi, H.; Katsuoka, F.; Shavit, J.A.; Engel, J.D.; Yamamoto, M. Positive or negative MARE-dependent transcriptional regulation is determined by the abundance of small maf proteins. Cell 2000, 103, 865–875.
- Yang, Y.; Cvekl, A. Large maf transcription factors: Cousins of AP-1 proteins and important regulators of cellular differentiation. Einstein J. Biol. Med. 2007, 23, 2–11.
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of maf and CNC families of transcription factors. Gene 2002, 294, 1–12.
- Ho, I.C.; Hodge, M.R.; Rooney, J.W.; Glimcher, L.H. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 1996, 85, 973–983.
- Blonska, M.; Joo, D.; Nurieva, R.I.; Zhao, X.; Chiao, P.; Sun, S.C.; Dong, C.; Lin, X. Activation of the transcription factor c-Maf in T cells is dependent on the CARMA1-IKKβ signaling cascade. Sci. Signal. 2013, 6, ra110.
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175.
- Sato, K.; Miyoshi, F.; Yokota, K.; Araki, Y.; Asanuma, Y.; Akiyama, Y.; Yoh, K.; Takahashi, S.; Aburatani, H.; Mimura, T. Marked induction of c-Maf protein during Th17 cell differentiation and its implication in memory Th cell development. J. Biol. Chem. 2011, 286, 14963–14971.
- Cao, S.; Liu, J.; Song, L.; Ma, X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J. Immunol. 2005, 174, 3484–3492.
- Kim, J.I.; Li, T.; Ho, I.C.; Grusby, M.J.; Glimcher, L.H. Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc. Natl. Acad. Sci. USA 1999, 96, 3781–3785.
- Kawauchi, S.; Takahashi, S.; Nakajima, O.; Ogino, H.; Morita, M.; Nishizawa, M.; Yasuda, K.; Yamamoto, M. Regulation of lens fiber cell differentiation by transcription factor c-Maf. J. Biol. Chem. 1999, 274, 19254–19260.
- Liu, F.Y.; Tang, X.C.; Deng, M.; Chen, P.; Ji, W.; Zhang, X.; Gong, L.; Woodward, Z.; Liu, J.; Zhang, L.; et al. The tumor suppressor p53 regulates c-Maf and Prox-1 to control lens differentiation. Curr. Mol. Med. 2012, 12, 917–928.
- Kase, S.; Yoshida, K.; Sakai, M.; Ohgami, K.; Shiratori, K.; Kitaichi, N.; Suzuki, Y.; Harada, T.; Ohno, S. Immunolocalization of cyclin D1 in the developing lens of c-maf -/- mice. Acta Histochem. 2006, 107, 469–472.
- Ring, B.Z.; Cordes, S.P.; Overbeek, P.A.; Barsh, G.S. Regulation of mouse lens fiber cell development and differentiation by the Maf gene. Development 2000, 127, 307–317.
- Nishikawa, K.; Nakashima, T.; Takeda, S.; Isogai, M.; Hamada, M.; Kimura, A.; Kodama, T.; Yamaguchi, A.; Owen, M.J.; Takahashi, S.; et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Investig. 2010, 120, 3455–3465.
- Kusakabe, M.; Hasegawa, K.; Hamada, M.; Nakamura, M.; Ohsumi, T.; Suzuki, H.; Tran, M.T.; Kudo, T.; Uchida, K.; Ninomiya, H.; et al. c-Maf plays a crucial role for the definitive erythropoiesis that accompanies erythroblastic island formation in the fetal liver. Blood 2011, 118, 1374–1385.
- Fujino, M.; Tagami, A.; Ojima, M.; Mizuno, S.; Abdellatif, A.M.; Kuno, A.; Takahashi, S. c-MAF deletion in adult C57BL/6J mice induces cataract formation and abnormal differentiation of lens fiber cells. Exp. Anim. 2020, 69, 242–249.
- Gómez-Salinero, J.M.; Izzo, F.; Lin, Y.; Houghton, S.; Itkin, T.; Geng, F.; Bram, Y.; Adelson, R.P.; Lu, T.M.; Inghirami, G.; et al. Specification of fetal liver endothelial progenitors to functional zonated adult sinusoids requires c-Maf induction. Cell Stem Cell 2022, 29, 593–609.e7.
- González-Loyola, A.; Bernier-Latmani, J.; Roci, I.; Wyss, T.; Langer, J.; Durot, S.; Munoz, O.; Prat-Luri, B.; Delorenzi, M.; Lutolf, M.P.; et al. c-MAF coordinates enterocyte zonation and nutrient uptake transcriptional programs. J. Exp. Med. 2022, 219, e20212418.
- Fujino, M.; Morito, N.; Hayashi, T.; Ojima, M.; Ishibashi, S.; Kuno, A.; Koshiba, S.; Yamagata, K.; Takahashi, S. Transcription factor c-Maf deletion improves streptozotocin-induced diabetic nephropathy by directly regulating Sglt2 and Glut2. JCI Insight2023, 8, e163306. https://doi.org/10.1172/jci.insight.163306.
- Jamieson, R.V.; Perveen, R.; Kerr, B.; Carette, M.; Yardley, J.; Heon, E.; Wirth, M.G.; van Heyningen, V.; Donnai, D.; Munier, F.; et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum. Mol. Genet. 2002, 11, 33–42.
- Jamieson, R.V.; Munier, F.; Balmer, A.; Farrar, N.; Perveen, R.; Black, G.C. Pulverulent cataract with variably associated microcornea and iris coloboma in a MAF mutation family. Br. J. Ophthalmol. 2003, 87, 411–412.
- Vanita, V.; Singh, D.; Robinson, P.N.; Sperling, K.; Singh, J.R. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am. J. Med. Genet. A 2006, 140, 558–566.
- Wende, H.; Lechner, S.G.; Cheret, C.; Bourane, S.; Kolanczyk, M.E.; Pattyn, A.; Reuter, K.; Munier, F.L.; Carroll, P.; Lewin, G.R.; et al. The transcription factor c-Maf controls touch receptor development and function. Science 2012, 335, 1373–1376.
- Niceta, M.; Stellacci, E.; Gripp, K.W.; Zampino, G.; Kousi, M.; Anselmi, M.; Traversa, A.; Ciolfi, A.; Stabley, D.; Bruselles, A.; et al. Mutations impairing GSK3-mediated MAF phosphorylation cause cataract, deafness, intellectual disability, seizures, and a Down syndrome-like facies. Am. J. Hum. Genet. 2015, 96, 816–825.
- Javadiyan, S.; Craig, J.E.; Sharma, S.; Lower, K.M.; Casey, T.; Haan, E.; Souzeau, E.; Burdon, K.P. Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures and hearing loss (Aymé-Gripp syndrome). BMC Med. Genet. 2017, 18, 52.
- Niceta, M.; Barbuti, D.; Gupta, N.; Ruggiero, C.; Tizzano, E.F.; Graul-Neumann, L.; Barresi, S.; Nishimura, G.; Valenzuela, I.; López-Grondona, F.; et al. Skeletal abnormalities are common features in Aymé-Gripp syndrome. Clin. Genet. 2020, 97, 362–369.
- Alkhunaizi, E.; Koenekoop, R.K.; Saint-Martin, C.; Russell, L. LMaternally inherited MAF variant associated with variable expression of Aymé-Gripp syndrome. Am. J. Med. Genet. A 2019, 179, 2233–2236.
- König, A.L.; Sabir, H.; Strizek, B.; Gembruch, U.; Herberg, U.; Bertrand, M.; Grasshoff, U.; Wiegand, G.; Wiechers, C.; Bernis, E.; et al. Isolated cytokine-enriched pericardial effusion: A likely key feature for Aymé-Gripp syndrome. Am. J. Med. Genet. A 2022, 188, 624–627.
- Fujino, M.; Ojima, M.; Ishibashi, S.; Mizuno, S.; Takahashi, S. Generation and mutational analysis of a transgenic murine model of the human MAF mutation. Am. J. Med. Genet. A 2023, 191, 1878–1888. https://doi.org/10.1002/ajmg.a.63220.
- Ogino, H.; Yasuda, K. Induction of lens differentiation by activation of a bZIP transcription factor, L-Maf. Science 1998, 280, 115–118.
- Kataoka, K.; Han, S.I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910.
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742.
- Matsuoka, T.A.; Zhao, L.; Artner, I.; Jarrett, H.W.; Friedman, D.; Means, A.; Stein, R. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol. Cell. Biol. 2003, 23, 6049–6062.
- Kataoka, K.; Shioda, S.; Ando, K.; Sakagami, K.; Handa, H.; Yasuda, K. Differentially expressed Maf family transcription factors, c-Maf and MafA, activate glucagon and insulin gene expression in pancreatic islet α- and β-cells. J. Mol. Endocrinol. 2004, 32, 9–20.
- Artner, I.; Le Lay, J.; Hang, Y.; Elghazi, L.; Schisler, J.C.; Henderson, E.; Sosa-Pineda, B.; Stein, R. MafB: An activator of the glucagon gene expressed in developing islet α- and β-cells. Diabetes 2006, 55, 297–304.
- Artner, I.; Blanchi, B.; Raum, J.C.; Guo, M.; Kaneko, T.; Cordes, S.; Sieweke, M.; Stein, R. MafB is required for islet β cell maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 3853–3858.
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab. 2011, 22, 364–373.
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976.
- Kato, T.; Shimano, H.; Yamamoto, T.; Yokoo, T.; Endo, Y.; Ishikawa, M.; Matsuzaka, T.; Nakagawa, Y.; Kumadaki, S.; Yahagi, N.; et al. Granuphilin is activated by SREBP-1c and involved in impaired insulin secretion in diabetic mice. Cell Metab. 2006, 4, 143–154.
- Iacovazzo, D.; Flanagan, S.E.; Walker, E.; Quezado, R.; de Sousa Barros, F.A.; Caswell, R.; Johnson, M.B.; Wakeling, M.; Brändle, M.; Guo, M.; et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2018, 115, 1027–1032.
- Walker, E.M.; Cha, J.; Tong, X.; Guo, M.; Liu, J.H.; Yu, S.; Iacovazzo, D.; Mauvais-Jarvis, F.; Flanagan, S.E.; Korbonits, M.; et al. Sex-biased islet β cell dysfunction is caused by the MODY MAFA S64F variant by inducing premature aging and senescence in males. Cell Rep. 2021, 37, 109813.
- Cordes, S.P.; Barsh, G.S. The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell 1994, 79, 1025–1034.
- Kataoka, K.; Fujiwara, K.T.; Noda, M.; Nishizawa, M. MafB, a new maf family transcription activator that can associate with maf and Fos but not with Jun. Mol. Cell. Biol. 1994, 14, 7581–7591.
- Sieweke, M.H.; Tekotte, H.; Frampton, J.; Graf, T. MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell 1996, 85, 49–60.
- Kelly, L.M.; Englmeier, U.; Lafon, I.; Sieweke, M.H.; Graf, T. MafB is an inducer of monocytic differentiation. EMBO J. 2000, 19, 1987–1997.
- Sadl, V.; Jin, F.; Yu, J.; Cui, S.; Holmyard, D.; Quaggin, S.; Barsh, G.; Cordes, S. The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev. Biol. 2002, 249, 16–29.
- Moriguchi, T.; Hamada, M.; Morito, N.; Terunuma, T.; Hasegawa, K.; Zhang, C.; Yokomizo, T.; Esaki, R.; Kuroda, E.; Yoh, K.; et al. MafB is essential for renal development and F4/80 expression in macrophages. Mol. Cell. Biol. 2006, 26, 5715–5727.
- Aziz, A.; Vanhille, L.; Mohideen, P.; Kelly, L.M.; Otto, C.; Bakri, Y.; Mossadegh, N.; Sarrazin, S.; Sieweke, M.H. Development of macrophages with altered actin organization in the absence of MafB. Mol. Cell. Biol. 2006, 26, 6808–6818.
- Kamitani-Kawamoto, A.; Hamada, M.; Moriguchi, T.; Miyai, M.; Saji, F.; Hatamura, I.; Nishikawa, K.; Takayanagi, H.; Hitoshi, S.; Ikenaka, K.; et al. MafB interacts with Gcm2 and regulates parathyroid hormone expression and parathyroid development. J. Bone Miner. Res. 2011, 26, 2463–2472.
- Sarrazin, S.; Mossadegh-Keller, N.; Fukao, T.; Aziz, A.; Mourcin, F.; Vanhille, L.; Kelly Modis, L.; Kastner, P.; Chan, S.; Duprez, E.; et al. MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 2009, 138, 300–313.
- Aziz, A.; Soucie, E.; Sarrazin, S.; Sieweke, M.H. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages. Science 2009, 326, 867–871.
- Sultana, D.A.; Tomita, S.; Hamada, M.; Iwanaga, Y.; Kitahama, Y.; Khang, N.V.; Hirai, S.; Ohigashi, I.; Nitta, S.; Amagai, T.; et al. Gene expression profile of the third pharyngeal pouch reveals role of mesenchymal MafB in embryonic thymus development. Blood 2009, 113, 2976–2987.
- Miyai, M.; Tanaka, Y.G.; Kamitani, A.; Hamada, M.; Takahashi, S.; Kataoka, K. c-Maf and MafB transcription factors are differentially expressed in Huxley’s and Henle’s layers of the inner root sheath of the hair follicle and regulate cuticle formation. J. Dermatol. Sci. 2010, 57, 178–182.
- Miyai, M.; Hamada, M.; Moriguchi, T.; Hiruma, J.; Kamitani-Kawamoto, A.; Watanabe, H.; Hara-Chikuma, M.; Takahashi, K.; Takahashi, S.; Kataoka, K. Transcription factor MafB coordinates epidermal keratinocyte differentiation. J. Investig. Dermatol. 2016, 136, 1848–1857.
- Suzuki, K.; Numata, T.; Suzuki, H.; Raga, D.D.; Ipulan, L.A.; Yokoyama, C.; Matsushita, S.; Hamada, M.; Nakagata, N.; Nishinakamura, R.; et al. Sexually dimorphic expression of Mafb regulates masculinization of the embryonic urethral formation. Proc. Natl. Acad. Sci. USA 2014, 111, 16407–16412.
- Dieterich, L.C.; Klein, S.; Mathelier, A.; Sliwa-Primorac, A.; Ma, Q.; Hong, Y.K.; Shin, J.W.; Hamada, M.; Lizio, M.; Itoh, M.; et al. DeepCAGE transcriptomics reveal an important role of the transcription factor MAFB in the lymphatic endothelium. Cell Rep. 2015, 13, 1493–1504.
- Tran, M.T.N.; Hamada, M.; Jeon, H.; Shiraishi, R.; Asano, K.; Hattori, M.; Nakamura, M.; Imamura, Y.; Tsunakawa, Y.; Fujii, R.; et al. MafB is a critical regulator of complement component C1q. Nat. Commun. 2017, 8, 1700.
- Shichita, T.; Ito, M.; Morita, R.; Komai, K.; Noguchi, Y.; Ooboshi, H.; Koshida, R.; Takahashi, S.; Kodama, T.; Yoshimura, A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 2017, 23, 723–732.
- Usui, T.; Morito, N.; Shawki, H.H.; Sato, Y.; Tsukaguchi, H.; Hamada, M.; Jeon, H.; Yadav, M.K.; Kuno, A.; Tsunakawa, Y.; et al. Transcription factor MafB in podocytes protects against the development of focal segmental glomerulosclerosis. Kidney Int. 2020, 98, 391–403.
- Katoh, M.C.; Jung, Y.; Ugboma, C.M.; Shimbo, M.; Kuno, A.; Basha, W.A.; Kudo, T.; Oishi, H.; Takahashi, S. MafB is critical for glucagon production and secretion in mouse pancreatic α cells in vivo. Mol. Cell. Biol. 2018, 38, e00504-17.
- Xiafukaiti, G.; Maimaiti, S.; Ogata, K.; Kuno, A.; Kudo, T.; Shawki, H.H.; Oishi, H.; Takahashi, S. MafB is important for pancreatic β-cell maintenance under a MafA-deficient condition. Mol. Cell. Biol. 2019, 39, e00080-19.
- Fujino, M.; Morito, N.; Hayashi, T.; Ojima, M.; Ishibashi, S.; Kuno, A.; Koshiba, S.; Yamagata, K.; Takahashi, S. Transcription factor c-Maf deletion improves streptozotocin-induced diabetic nephropathy by directly regulating Sglt2 and Glut2. JCI Insight 2023, 8, e163306.
- Zankl, A.; Duncan, E.L.; Leo, P.J.; Clark, G.R.; Glazov, E.A.; Addor, M.C.; Herlin, T.; Kim, C.A.; Leheup, B.P.; McGill, J.; et al. Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am. J. Hum. Genet. 2012, 90, 494–501.
- Park, J.G.; Tischfield, M.A.; Nugent, A.A.; Cheng, L.; Di Gioia, S.A.; Chan, W.M.; Maconachie, G.; Bosley, T.M.; Summers, C.G.; Hunter, D.G.; et al. Loss of MAFB function in humans and mice causes Duane syndrome, aberrant extraocular muscle innervation, and inner-ear defects. Am. J. Hum. Genet. 2016, 98, 1220–1227.
- Sato, Y.; Tsukaguchi, H.; Morita, H.; Higasa, K.; Tran, M.T.N.; Hamada, M.; Usui, T.; Morito, N.; Horita, S.; Hayashi, T.; et al. A mutation in transcription factor MAFB causes Focal Segmental glomerulosclerosis with Duane retraction syndrome. Kidney Int. 2018, 94, 396–407.
- Tsunakawa, Y.; Hamada, M.; Matsunaga, Y.; Fuseya, S.; Jeon, H.; Wakimoto, Y.; Usui, T.; Kanai, M.; Mizuno, S.; Morito, N.; et al. Mice harboring an MCTO mutation exhibit renal failure resembling nephropathy in human patients. Exp. Anim. 2019, 68, 103–111.
- Mehawej, C.; Courcet, J.B.; Baujat, G.; Mouy, R.; Gérard, M.; Landru, I.; Gosselin, M.; Koehrer, P.; Mousson, C.; Breton, S.; et al. The identification of MAFB mutations in eight patients with multicentric carpo-tarsal osteolysis supports genetic homogeneity but clinical variability. Am. J. Med. Genet. A 2013, 161, 3023–3029.
This entry is offline, you can click here to edit this entry!