Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Pulmonary hypertension (PH) is a severe vascular complication of connective tissue diseases (CTD). Patients with CTD may develop PH belonging to diverse groups: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease, (3) secondary PH due to lung disease and/or hypoxia and (4) chronic thromboembolic pulmonary hypertension (CTEPH). PAH most often develops in systemic scleroderma (SSc), mostly in its limited variant. PAH-CTD is a progressive disease characterized by poor prognosis. Therefore, early diagnosis should be established.
- PAH CTD
- novel drugs
- therapy
- treatment
- prognosis
1. Introduction
Pulmonary hypertension (PH) is a vascular complication of connective tissue diseases (CTD), where, in addition to the symptoms of CTD with the involvement of many systems and organs on an autoimmune basis, pulmonary pressure is increased, and which, if left untreated, leads to severe right ventricular failure and death. Moreover, patients with CTD may develop PH belonging to different groups: (i) pulmonary arterial hypertension (PAH), (ii) PH due to left heart disease, (iii) PH secondary to lung disease and/or hypoxia and (iv) chronic thromboembolic pulmonary hypertension (CTEPH), especially in the setting of antiphospholipid syndrome (Figure 1) [1]. Pre-capillary PH is diagnosed by right-sided cardiac catheterization (RHC) when the following hemodynamic criteria are met: mean pulmonary arterial pressure (mPAP) > 20 mmHg, pulmonary vascular resistance (PVR) > 2.0 Woods units and pre-capillary wedge pressure (PCWP) ≤ 15 mmHg. PAH most often develops in systemic scleroderma (SSc), mainly in its limited variant [2][3][4]. The prevalence of pre-capillary PH (PAH and PH secondary to lung disease) in a large group of patients with SSc is estimated at 5–19% [2][3]. Other CTD associated with PAH development include systemic lupus erythematosus (SLE) [4][5][6], mixed CTD [4], rheumatoid arthritis [7], dermatomyositis [8] and Sjögren’s syndrome [9]. A specific treatment for PAH-CTD is currently available and recommended: prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase-five inhibitors (PDE5i: sildenafil, tadalafil), endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan) [10]. Until now, it was thought that the treatment of arterial pulmonary hypertension in patients with PAH-CTD is less effective than in patients without CTD [11][12][13].
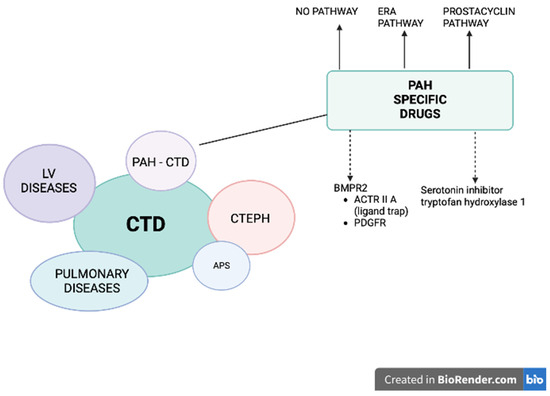
Figure 1. Patients with connective tissue diseases (CTD) may develop pulmonary hypertension (PH) belonging to different groups: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease, (3) PH secondary to lung disease and/or hypoxia (CTD patients mostly develop interstitial lung disease), and (4) chronic thromboembolic pulmonary hypertension (CTEPH), especially in the setting of antiphospholipid syndrome (APS). A specific treatment for PAH-CTD is currently available and recommended (solid arrows): prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase-5 inhibitors (PDE5i: sildenafil, tadalafil) and endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan). Two other pathways are under intensive investigation (dashed arrows): (1) affecting BMPR2: trapping the ligands of TGF-β and reducing the activity of ACTRIIA (sotatercept, KER-012) or inhibiting PDGFR (imatinib, seralutinib), (2) inhibiting the serotonin pathway by blocking the serotonin-producing enzyme tryptophan hydroxylase 1 (rodatristat ethyl).
2. Diagnosis and Risk Assessment
It has been proven that asymptomatic patients with SSc should undergo screening to diagnose PAH as soon as possible and initiate specific treatment [14]. In 2011, Humbert et al. showed that patients with SSc undergoing screening with echocardiography compared to routine clinical practice are significantly more often diagnosed at an early stage of PAH in WHO FC I and II. It is now believed that all patients with SSc should undergo extended screening, preferably using a validated DETECT scale. This complex scale is characterized by higher specificity compared to echocardiography alone, leading to a faster diagnosis of PAH [15]. The DETECT scale includes forced vital capacity (FVC) percent predictive value, diffusion lung capacity for carbon monoxide (DLCO) percent predictive value, telangiectasias, anti-centromere antibody, N-terminal brain natriuretic propeptide (NT-proBNP), serum urate and right axis deviation on ECG. The next diagnostic step, according to the DETECT scale, is echocardiography, and the last is right-sided cardiac catheterization (RHC) [2].
Death risk assessment is performed regularly during diagnosis and follow-ups every four to six months. According to the ESC guidelines, initial risk evaluation should include clinical, laboratory and echocardiographic evaluation, exercise capacity (six-minute walking test [6MWT]), heart pulmonary exercise test), hemodynamic parameters derived from RHC, as well as magnetic resonance (MRI) measurements [10]. Based on these parameters, patients are initially divided on a three-strata scale into high risk, intermediate risk and low risk of death. During scheduled visits, the highest predictive value is 6MWT, NT-proBNP or BNP, and the WHO-FC.
The latest data from the COMPERA [16] and French registries [17] confirmed the highest mortality in the long-term observation of patients with PAH-CTD at high risk. In the Australian prospective study, independent predictive mortality factors in PAH-CTD patients were of older age, higher mPAP at PAH diagnosis, worse WHO FC and digital ulcers. Interestingly, the 6MWD was not predictive of mortality [18]. It can be explained by the observation that patients with PAH-CTD often have limited physical activity resulting from musculoskeletal involvement. Undoubtedly, the presence and severity of interstitial lung disease (ILD) significantly worsens the prognosis of patients with SSc. Moreover, sub-analysis has shown that patients with pre-capillary PH and extensive ILD have a worse prognosis compared to the subgroup of patients with mild ILD or without the ILD [12][19].
3. Therapy
Starting therapy at an early stage of PAH significantly improves the patients’ prognosis. In the French registry, three-year survival in patients with SSc was 80% when treatment was initiated in the NYHA (New York Heart Association)/WHO functional class II, 72% in stage III and 30% in IV [20].
The treatment of PAH-CTD involves drugs affecting several pathways/mechanisms, and the initial therapy depends on the severity of the disease. According to the ESC guidelines, like patients with idiopathic PAH, patients with PAH-CTD in the high-risk group should start PAH pharmacotherapy with a combination of triple therapy containing ERA, PDE5i and parenteral (s.c., i.v., inhaled) prostacyclin [10]. Patients with low- or intermediate-risk should start with dual combination therapy containing ERA, preferred macitentan or ambrisentan and PDE5i or riociguat. An initially implemented triple-combination treatment approach, including i.v./s.c. prostanoids should also be considered in intermediate risk with severe hemodynamic impairment, e.g., mean right atrial pressure (mRAP) ≥ 20 mmHg, cardiac index (CI) < 2.0 L/min/m2, stroke volume index < 31 mL/m2, PVR ≥ 12 Woods units. As comorbidities are common in PAH-CTD, the initial treatment of patients with cardiopulmonary risk factors (advanced age, obesity, atrial fibrillation, history of hypertension, coronary artery disease and/or low DLCO) should be considered as monotherapy with PDE5i or ERA.
The treatment, response to the therapy and management of patients with PAH-CTD should be provided by referral centers.
Furthermore, when triple therapy including parenteral prostacyclin analogue is initiated, it is recommended to refer these patients to specialized surgical centers for eligibility for bilateral lung transplantation. Lung transplantation is not contraindicated in patients with CTD-PAH. However, evaluating other potentially involved organs, especially the digestive system, kidneys, heart and skin, must be considered [21][22].
Another interventional treatment in advanced PAH includes balloon atrial septostomy (BAS) [23] by performing an interatrial shunt, leading to decompression of the right atrium and ventricle and increased systemic blood flow at the expense of arterial oxygen saturation. BAS should be performed in the experienced PAH centers as a bridge to lung transplantation or ultimate therapy, preferably with atrial flow regulator (AFR) [24]. Other interventional treatments are under clinical investigation: denervation of pulmonary arteries (PADN) by using the radiofrequency ablation [25] or intravascular ultrasound catheter [26]. The latter was characterized by good safety measures and showed improvement in 6MWT and reduced PVR.
Analysis of subgroups of patients with PAH-CTD and PAH-SSc from randomized trials with sildenafil, tadalafil, bosentan, ambrisentan, macitentan, treprostinil, iloprost, riociguat and selexipag showed a beneficial effect. These subsections present alternative pharmacological targets and their efficacy in CTD patients with PAH.
Some of the studies investigating PAH-specific drugs have shown that the effect of treatment was worse in patients with CTD-PAH than in patients with idiopathic PAH [27]. However, the primary endpoint in these studies was most often the 6MWT. Nevertheless, the results of a 2021 meta-analysis of 11 randomized trials (n-4329, n-1267 CTD-PAH) and 19 registries (n-9739, n-4008 CTD-PAH) with at least 30-person subgroups with CTD-PAH from 2000–2019 showed a beneficial effect of modern PAH-specific treatment in both CTD-PAH and PAH groups. The meta-analysis of randomized trials in which the endpoint was composite (time to event mortality/morbidity including death, worsening, hospitalization for prostacyclin, lung transplantation or septostomy) showed the same 36% reduction in the risk of incident mortality/morbidity in patients treated with PAH-specific therapy in comparison with placebo in both the overall group and the subgroup with CTD-PAH. However, at baseline, both in the RCTs and in the registries, patients with CTD-PAH had a lower mean 6MWT and were older compared to the overall PAH population. In addition, PAH-CTD patients had a higher mortality rate when compared with other groups of patients with PAH (three-year survival was 62% in patients with CTD-PAH and 72% in all patients) [28]. Nevertheless, the three-year survival of patients with CTD-PAH after 2010 was better than before 2010 (73% vs. 65%, respectively) [29]. The AMBITION study (whose primary endpoint was clinical worsening) showed that in all patients, regardless of PAH etiology, initial composite treatment with tadalafil and bosentan was preferable to initial monotherapy. These results were proved in a post hoc analysis exclusively assessing patients with PAH-CTD (187 patients with PAH-CTD, of whom 118 had SSc-PAH) [30].
4. Conclusions
PAH-CTD is a progressive disease that must be detected early (patients with SSc should be screened annually), and composite drug therapy is recommended to be implemented as early as possible [31]. Despite a serious prognosis, PAH-CTD can be effectively treated with PAH-specific drugs. The number of drugs (monotherapy, dual or triple therapy) initiated in PAH-CTD depends on the risk stratification and presence of comorbidities at the time of the diagnosis. There are several intensively investigated novel drugs, from which the most promising are agents affecting BMPR2 signaling (sotatercept), PDGFR (imatinib, seralutinib) and the tryptophan pathway (rodatristat ethyl).
This entry is adapted from the peer-reviewed paper 10.3390/ph16091252
References
- Fayed, H.; Coghlan, J.G. Pulmonary Hypertension Associated with Connective Tissue Disease. Semin. Respir. Crit. Care Med. 2019, 40, 173–183.
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 37, 1340–1349.
- Avouac, J.; Airò, P.; Meune, C.; Beretta, L.; Dieude, P.; Caramaschi, P.; Tiev, K.; Cappelli, S.; Diot, E.; Vacca, A.; et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and meta-analysis of 5 studies. J. Rheumatol. 2010, 37, 2290–2298.
- Jais, X.; Launay, D.; Yaici, A.; Le Pavec, J.; Tchérakian, C.; Sitbon, O.; Simonneau, G.; Humbert, M. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: A retrospective analysis of twenty-three cases. Arthritis Rheumatol. 2008, 58, 521–531.
- Hachulla, E.; Jais, X.; Cinquetti, G.; Clerson, P.; Rottat, L.; Launay, D.; Cottin, V.; Habib, G.; Prevot, G.; Chabanne, C.; et al. French Collaborators Recruiting Members. Pulmonary Arterial Hypertension Associated With Systemic Lupus Erythematosus: Results From the French Pulmonary Hypertension Registry. Chest 2018, 153, 143–151.
- Qian, J.; Li, M.; Zhang, X.; Wang, Q.; Zhao, J.; Tian, Z.; Wei, W.; Zuo, X.; Zhang, M.; Zhu, P.; et al. Long-term prognosis of patients with systemic lupus erythematosus-associated pulmonary arterial hypertension: CSTAR-PAH cohort study. Eur. Respir. J. 2019, 53, 1800081.
- Kopeć, G.; Kurzyna, M.; Mroczek, E.; Chrzanowski, Ł.; Mularek-Kubzdela, T.; Skoczylas, I.; Kuśmierczyk, B.; Pruszczyk, P.; Błaszczak, P.; Lewicka, E.; et al. Characterization of Patients with Pulmonary Arterial Hypertension: Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 173.
- Sanges, S.; Yelnik, C.M.; Sitbon, O.; Benveniste, O.; Mariampillai, K.; Phillips-Houlbracq, M.; Pison, C.; Deligny, C.; Inamo, J.; Cottin, V.; et al. Pulmonary arterial hypertension in idiopathic inflammatory myopathies: Data from the French pulmonary hypertension registry and review of the literature. Medicine 2016, 95, e4911.
- Wang, J.; Li, M.; Wang, Q.; Zhang, X.; Qian, J.; Zhao, J.; Xu, D.; Tian, Z.; Wei, W.; Zuo, X.; et al. Pulmonary arterial hypertension associated with primary Sjögren’s syndrome: A multicentre cohort study from China. Eur. Respir. J. 2020, 56, 1902157.
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur. Heart J. 2022, 43, 3618–3731.
- Launay, D.; Montani, D.; Hassoun, P.M.; Cottin, V.; Le Pavec, J.; Clerson, P.; Sitbon, O.; Jaïs, X.; Savale, L.; Weatherald, J.; et al. Clinical phenotypes and survival of pre-capillary pulmonary hypertension in systemic sclerosis. PLoS ONE 2018, 13, e0197112.
- Launay, D.; Sitbon, O.; Hachulla, E.; Mouthon, L.; Gressin, V.; Rottat, L.; Clerson, P.; Cordier, J.F.; Simonneau, G.; Humbert, M. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann. Rheum. Dis. 2013, 72, 1940–1946.
- Ramjug, S.; Hussain, N.; Hurdman, J.; Billings, C.; Charalampopoulos, A.; Elliot, C.A.; Kiely, D.G.; Sabroe, I.; Rajaram, S.; Swift, A.J.; et al. Idiopathic and Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: A Comparison of Demographic, Hemodynamic, and MRI Characteristics and Outcomes. Chest 2017, 152, 92–102.
- Humbert, M.; Yaici, A.; de Groote, P.; Montani, D.; Sitbon, O.; Launay, D.; Gressin, V.; Guillevin, L.; Clerson, P.; Simonneau, G.; et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: Clinical characteristics at diagnosis and long-term survival. Arthritis Rheumatol. 2011, 63, 3522–3530.
- Young, A.; Moles, V.M.; Jaafar, S.; Visovatti, S.; Huang, S.; Vummidi, D.; Nagaraja, V.; McLaughlin, V.; Khanna, D. Performance of the DETECT Algorithm for Pulmonary Hypertension Screening in a Systemic Sclerosis Cohort. Arthritis Rheumatol. 2021, 73, 1731–1737.
- Hoeper, M.M.; Pausch, C.; Grünig, E.; Staehler, G.; Huscher, D.; Pittrow, D.; Olsson, K.M.; Vizza, C.D.; Gall, H.; Distler, O.; et al. Temporal trends in pulmonary arterial hypertension: Results from the COMPERA registry. Eur. Respir. J. 2022, 59, 2102024.
- Boucly, A.; Weatherald, J.; Savale, L.; de Groote, P.; Cottin, V.; Prévot, G.; Chaouat, A.; Picard, F.; Horeau-Langlard, D.; Bourdin, A.; et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur. Respir. J. 2022, 59, 2102419.
- Morrisroe, K.; Stevens, W.; Huq, M.; Prior, D.; Sahhar, J.; Ngian, G.S.; Celermajer, D.; Zochling, J.; Proudman, S.; Nikpour, M.; et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res. Ther. 2017, 19, 122.
- Zanatta, E.; Marra, M.P.; Famoso, G.; Balestro, E.; Giraudo, C.; Calabrese, F.; Rea, F.; Doria, A. The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease. Pharmaceuticals 2022, 15, 1042.
- Hachulla, E.; Launay, D.; Yaici, A.; Berezne, A.; de Groote, P.; Sitbon, O.; Mouthon, L.; Guillevin, L.; Hatron, P.Y.; Simonneau, G.; et al. Pulmonary arterial hypertension associated with systemic sclerosis in patients with functional class II dyspnea: Mild symptoms but severe outcome. Rheumatology 2010, 49, 940–944.
- Gadre, S.K.; Minai, O.A.; Wang, X.F.; Zhang, Q.; Budev, M.; Tonelli, A.R. Lung or heart-lung transplant in pulmonary arterial hypertension: What is the impact of systemic sclerosis? Exp. Clin. Transplant. 2017, 15, 676–684.
- Mularek-Kubzdela, T.; Wojnarski, J.; Kamiński, K.; Ochman, M.; Kasprzak, J.; Stącel, T.; Kurzyna, M.; Karolak, W.; Mroczek, E.; Kopeć, G.; et al. Lung transplantation in patients with pulmonary arterial hypertension: The opinion of the Polish Cardiac Society Working Group on Pulmonary Circulation. Kardiol. Pol. 2022, 80, 1169–1181.
- Khan, M.S.; Memon, M.M.; Amin, E.; Yamani, N.; Khan, S.U.; Figueredo, V.M.; Deo, S.; Rich, J.D.; Benza, R.L.; Krasuski, R.A. Use of Balloon Atrial Septostomy in Patients With Advanced Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Chest 2019, 156, 53–63.
- Kopeć, G.; Araszkiewicz, A.; Magoń, W.; Stępniewski, J.; Sławek-Szmyt, S.; Janus, M.; Skoczylas, I.; Gąsior, Z.; Wilczek, Ł.; Komar, M.; et al. The results of atrial flow regulator implantation in pulmonary arterial hypertension patients with severe heart failure despite maximal medical therapy. Kardiol. Polska 2023. epub ahead of print.
- Chen, S.L.; Zhang, F.F.; Xu, J.; Xie, D.J.; Zhou, L.; Nguyen, T.; Stone, G.W. Pulmonary artery denervation to treat pulmonary arterial hypertension: The single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). J. Am. Coll. Cardiol. 2013, 62, 1092–1100.
- Rothman, A.M.K.; Vachiery, J.L.; Howard, L.S.; Mikhail, G.W.; Lang, I.M.; Jonas, M.; Kiely, D.G.; Shav, D.; Shabtay, O.; Avriel, A.; et al. Intravascular Ultrasound Pulmonary Artery Denervation to Treat Pulmonary Arterial Hypertension (TROPHY1): Multicenter, Early Feasibility Study. JACC Cardiovasc. Interv. 2020, 13, 989–999.
- Almaaitah, S.; Highland, K.B.; Tonelli, A.R. Management of pulmonary arterial hypertension in patients with systemic sclerosis. Integr. Blood Press. Control 2020, 13, 15–29.
- Zhang, C.; Wang, X.; Zhang, H.; Yao, C.; Pan, H.; Guo, Y.; Fan, K.; Jing, S. Therapeutic Monoclonal Antibody Antagonizing Endothelin Receptor A for Pulmonary Arterial Hypertension. J. Pharmacol. Exp. Ther. 2019, 370, 5461.
- Khanna, D.; Zhao, C.; Saggar, R.; Khanna, D.; Zhao, C.; Saggar, R.; Mathai, S.C.; Chung, L.; Coghlan, J.G.; Shah, M.; et al. Long-Term outcomes in patients with connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era: Meta-analyses of randomized, controlled trials and observational registries. Arthritis Rheumatol. 2021, 73, 837–847.
- Kuwana, M.; Blair, C.; Takahashi, T.; Langley, J.; Coghlan, J.G. Initial combination therapy of ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) in the modified intention-to-treat population of the AMBITION study: Post hoc analysis. Ann. Rheum. Dis. 2020, 79, 626–634.
- Mularek-Kubzdela, T.; Ciurzyński, M.; Kowal Bielecka, O.; Kasprzak, J.D.; Kopeć, G.; Mizia-Stec, K.; Mroczek, E.; Lewicka, E.; Skoczylas, I.; Grabka, M.; et al. An expert opinion of the Polish Cardiac Society Working Group on Pulmonary Circulation and the Polish Society for Rheumatology on the diagnosis and treatment of pulmonary hypertension in patients with connective tissue disease. Kardiol. Pol. 2021, 79, 917–929.
This entry is offline, you can click here to edit this entry!