Cancer, also known as malignant tumour or neoplasm, is a leading cause of death worldwide. One distinct feature from normal cells is that cancerous cells often overexpress protein on the cell membrane—for instance, the overexpression of human epidermal growth factor receptor 2. The expression of a specific protein on the cancerous cell surface acts as a marker that differentiates the normal cell and facilitates the recognition of cancerous cells. An emerging anticancer treatment, Antibody–Drug Conjugates (ADCs), utilises this unique feature to kill cancerous cells. ADCs consist of an antibody linked with a cytotoxic payload, mainly targeting the antigen found on cancerous cells. This design can increase the specificity in delivering the cytotoxin to the drug target, thus increasing the drug efficacy and reducing the side effect of cancer treatment due to off-target toxicities.
1. Introduction
The discovery of the “Magic Bullet Theory” by a German Scientist, Paul Ehrlich, marked the targeted therapy revolution. He espoused the theory to describe his vision, where a chemical binds selectively to targeted microorganisms [
1]. Ehrlich eventually realised this concept by successfully treating syphilis with a highly selective drug towards bacteria that does not harm the neighbouring healthy cells [
2]. His outstanding achievement has had a significant positive impact on the healthcare system in which scientists develop drugs that could specifically target the cells in the body system.
The concept of the “magic bullet” has to some extent been realised by the development of antibody–drug conjugates (ADCs), particularly in the field of oncology. ADCs’ historical revolution has answered the crucial pharmaceutical concern of all oncologists, the need for anticancer treatment to target tumour cells efficiently along with high precision and specificity. The tremendous advancement of monoclonal antibody technology has allowed various cytotoxic agents such as cytotoxic drugs, immunotoxins, and radiopharmaceuticals to be conjugated to the bullet, resulting in high selectivity and targetability on the specific antigen expressed on cancerous cells. ADCs were proposed to reduce non-specific toxicities by altering their signalling pathways towards a therapeutic outcome or directing naturally acquired immune responses towards the tumour cells [
3]. In recent years, new ADCs have been under clinical development, marking the success of this highly potential therapeutic agent in chemotherapy (
Figure 1) [
4]. Drago et al. (2021) have been very insightful in ADC design and maximising the potential of ADCs for cancer therapy [
5].
2. Principle of Antibody–Drug Conjugate
ADC comprises a tumour antigen-specific antibody, a potent cytotoxic drug and a stable chemical linker that joins the antibody to the cytotoxic drug [
10]. The physicochemical properties of respective components in an ADC are summarised in
Table 1.
Table 1. Summary of the physicochemical properties of respective components in an ADC.
| Components of ADC |
| Antibody |
Subunit |
Function |
| Antigen-binding Fragment (Fab) |
Mediate antigen recognition expressed on tumour cells. |
| Constant Fragment (Fc) |
Facilitate binding of Fab to immune cells. |
| Linker |
Type |
Mechanism of Drug Release |
| Cleavable |
Cleavage depends on the physiological environment.
-
Acid labile linker is cleaved at low pH, e.g., Gemtuzumab Ozogamicin.
-
Protease cleavable linker is cleaved through proteolysis, e.g., Brentuximab Vedotin.
-
Disulphide linker is cleaved by high intracellular glutathione concentrations, e.g., Mirvetuximab Soravtansine.
|
| Non-cleavable |
It depends on lysosomal proteolytic degradation and requires optimal trafficking to lysosome. Thioether linker is cleaved by proteases in the cytosolic milieu, e.g., Trastuzumab Emtansine (T–DM1). |
| Cytotoxic Payload |
Target Site |
Examples of Payload with
Mechanism of Action |
| DNA |
-
Calicheamicins induce double-strand DNA breaks.
-
Duocarmycin and Pyrrolobenzodiazepines (PBD) induce DNA alkylation by binding to A–T rich regions and guanine residues, respectively.
|
| Tubulin |
-
Auristatins and derivatives of Maytansine inhibit polymerisation of the microtubule and cell cycle arrest at the G2/M phase.
-
Auristatin monomethyl auristatin E (MMAE) in Brentuximab vedotin and glembatumumab vedotin, Monomethyl auristatin F (MMAF) in Depatuximab mafodotin, a derivative of Maytansine (DM1) in trastuzumab emtansine.
|
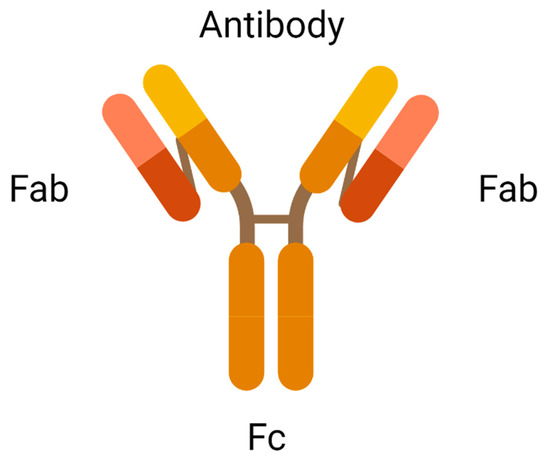
2.1. Antibody
An antibody is the most crucial element in an ADC due to its role in recognising the cancer surface marker. Therefore, an excellent antibody used in ADC should be stable during circulation in the bloodstream, less immunogenic, highly specific for the antigen, and can be internalised by the cancer cells. There are two antigen-binding fragments known as Fabs to recognise antigens expressed on cancer cells over healthy cells. A constant fragment, Fc mediates the interaction of antibodies with immune cells such as lymphocytes and B cells (
Figure 2). A binding domain on the Fc receptor binds to the neonatal Fc receptor (FcRn) that regulates serum protein half-life [
11]. Selection of the appropriate antibody subtype or technological engineering of the Fc domain, antigen affinity, the immune function of ADC, and biodistribution will influence the immune function of an ADC. In addition to the intrinsic cytotoxicity of drugs, the antibody on ADC mediates immune functions that are naturally acquired in patients through the activation of complement immune effector cells and signalling components, resulting in the migration of immune effector cells to the targeted sites for tumour-killing effects.
2.2. Cytotoxic Payload
Cytotoxic payload is the effector component in ADC. The ideal candidate of an anticancer drug used in ADC should be highly potent with half-maximal inhibitory concentration (IC50) in the subnanomolar range to achieve desired therapeutic activities. Subnanomolar potency is crucial because only a few payloads can be conjugated to the targeting antibodies. Low potency of payloads results in a treatment failure. Due to the criteria of subnanomolar potency, many highly toxic drugs that were abandoned in the laboratory have now been reviewed as potential payloads in the development of ADCs. Common intracellular targets of ADC in tumour cells are DNA and Tubulin. The inhibition of these targets blocks the mitotic phase of tumour cells, slowing cell growth, and ultimately inducing apoptotic cell death [
12].
2.3. Linker
The chemical linker in ADC conjoins cytotoxic payload to the antibody, stabilising the circulating ADC in the bloodstream upon administration. The functional group of linkers and the conjugation site determine the performance of ADCs in terms of circulating half-life, pharmacokinetics and pharmacodynamic profiles, and therapeutic window [
14]. Disulfide, thioether, and hydrazone functional groups combine antibodies to the cytotoxic agent through intermolecular interactions. Currently, available linkers based on mechanisms of payload release are classified as cleavable or non-cleavable. Cleavable linkers depend on the physiological environment in the systemic circulation, such as pH or proteases available to release cytotoxic payload from the carrier. For example, the acid–labile linker in Gemtuzumab Ozogamicin is cleaved under low pH conditions. In contrast, protease cleavable linker in Brentuximab Vedotin is cleaved by proteases such as Cathepsin-B and plasmin. The cleavage of disulfide linkers in mirvetuximab soravtansine depends on high glutathione concentrations [
15]. Non-cleavable linkers form non-reducible covalent bonds with amino acids on the antibodies. Unlike cleavable linkers, non-cleavable linkers are not affected by the physiological environment and are more stable in blood circulation [
16].
2.4. Mechanism of Action of ADC
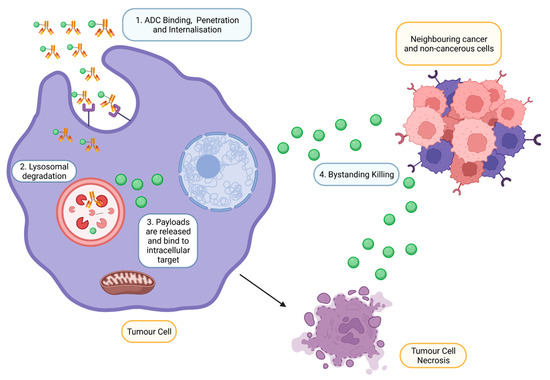
Upon intravenous administration, ADC circulates in the bloodstream and binds to an antigen explicitly expressed by targeted tumour cells (
Figure 3) [
18]. Following internalisation, ATP-dependent proton pumps in lysosomes to create an acidic environment to facilitate the degradation of cleavable ADCs by proteases such as plasmin and cathepsin-B, releasing cytotoxic drugs from ADC complexes [
11]. In addition, lysosomal degradation, proteolytic cleavage of acid–labile linker, protease cleavable linker, and disulfide linker by the physiological environment occurs in the early or late endosomes [
19]. The released cytotoxic drugs in the free circulating state resulted in the binding to intracellular targets, causing apoptosis via DNA intercalation or inhibition of microtubule polymerisation. As tumour cells lyse, free cytotoxic drugs are released and progress to bystander killing of neighbouring cancerous and non-cancerous cells via passive diffusion [
20]. Apart from direct cytotoxicity, ADC-mediated effector functions include activating complement systems and infiltration of immune cells to localise at the targeted tumour via several mechanisms such as ADCP and ADCC [
21].
Figure 3. Mechanism of action of ADCs at targeted tumour cells. ADCs are rapidly internalised and degraded in lysosomes upon binding to the antigen on the tumour. Payloads are then released into the cytoplasm, bind to the intracellular target and induce cell death. However, payloads can also exhibit bystander effects by diffusing out from the cells into adjacent cells (Created with
https://www.biorender.com/) (accessed on 2 December 2022).
3. Antibody–Drug Conjugate Approved by FDA
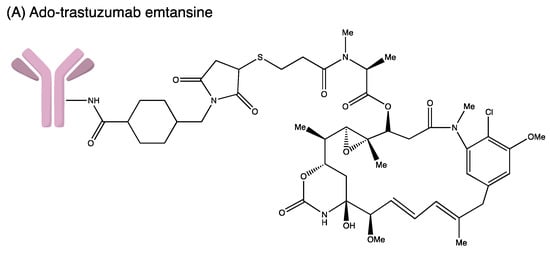
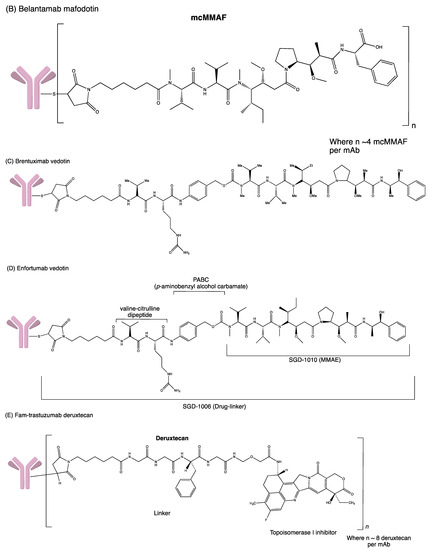
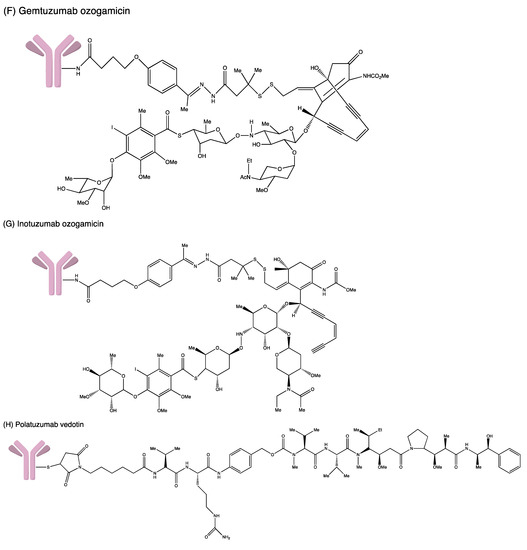
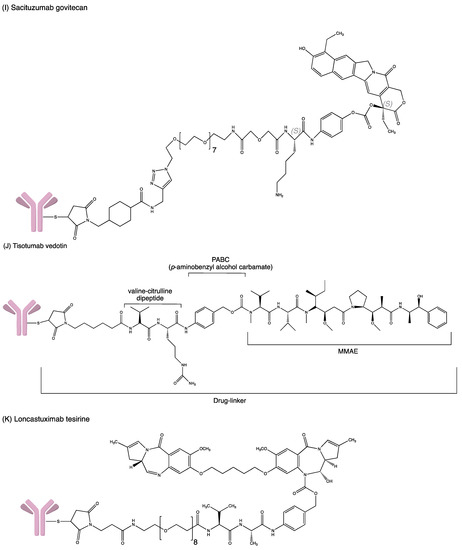
Developing an ADC is not easy as the process involves complicated bioengineering, chemistry and pharmacology. Many ADCs have recently received FDA-accelerated approval to treat severe malignancies in the hospital setting despite the challenges and hurdles (Figure 4). The FDA-approved ADCs are highlighted and updated accordingly in Table 2.
Figure 4. Structures of (A) ado-trastuzumab emtansine. (B) belantamab mafodotin. (C) brentuximab vedotin. (D) enfortumab vedotin. (E) fam-trastuzumab deruxtecan. (F) gemtuzumab ozogamicin. (G) inotuzumab ozogamicin (H) polatuzumab vedotin. (I) sacituzumab govitecan. (J) tisotumab vedotin. (K) loncastuximab tesirine.
Table 2. FDA-approved Antibody–Drug Conjugates.
| ADC |
Antibody |
Target Antigen |
Linker |
Cytotoxic Payload |
Indication |
Year of Approval |
| Gemtuzumab ozogamicin |
Humanised IgG4 |
CD33 |
Acid–labile hydrazone |
Calicheamicin |
Relapse or refractory AML and newly diagnosed CD33+ AML |
Approved in 2000, withdrawn in 2010 but then reapproved in 2017 for relapsed/refractory malignancies
FDA approved for newly diagnosed CD33+ AML in ≥1-month paediatric patient |
| Brentuximab vedotin |
Chimeric IgG1 |
CD30 |
Protease-cleavable dipeptide |
MMAE |
HL, ALCL and different subtypes of T-cell lymphomas |
FDA accelerated approval in 2011
In 2018, FDA approved the treatment of previously untreated stage III-IV HL and previously untreated ALCL and other CD30+ peripheral T-cell lymphomas. |
| Ado-trastuzumab emtansine |
Humanised IgG1 |
HER2 |
SMCC |
DM1 |
Metastatic HER2+ breast cancer, previously treated with trastuzumab and a taxane and as adjuvant treatment for HER2+ early breast cancer with the residual invasive disease after neoadjuvant taxane and trastuzumab |
FDA approved in 2013
FDA approved for adjuvant treatment in 2019 |
| Inotuzumab Ozogamicin |
Humanised IgG4 |
CD22 |
Acid–labile hydrazone |
Calicheamicin |
Relapsed or refractory B-cell precursor ALL |
FDA approved in February 2017 |
| Monotherapy treatment of relapsed or refractory CD22-positive B-cell precursor |
EMA approved in June 2017 |
| Polatuzumab vedotin |
Humanised IgG1 |
CD79b |
Cleavable dipeptide |
MMAE |
Relapsed or refractory diffuse large B-cell lymphoma |
FDA accelerated approval in 2019 |
| Enfortumab vedotin |
Fully human IgG1 |
Nectin 4 |
Protease-cleavable dipeptide (Val–Cit) linker |
MMAE |
Locally advanced or metastatic urothelial cancer in adult patients who received prior treatment with a PD-1/L1 inhibitor and platinum-based chemotherapy in neoadjuvant/adjuvant setting |
FDA accelerated approved in 2019 |
| Trastuzumab deruxtecan |
Humanised IgG1 |
HER2/ERB2 |
Protease-cleavable tetra-peptide (Gly–Gly–Phe–Gly) linker |
DXd |
Unresectable locally advanced or metastatic HER2+ breast cancer, previously treated with trastuzumab and a taxane, adjuvant treatment for HER2+ early breast cancer with the residual invasive disease after neoadjuvant taxane and trastuzumab |
FDA accelerated approved in 2019 |
| Sacituzumab govitecan |
Humanised IgG1 |
TROP-2 |
Acid–labile ester (CL2 linker) |
SN-38 |
Triple-negative breast cancer, urothelial and other cancers |
FDA accelerated approval in April 2020 for mTNBC |
| FDA regular approval for in April 2021 TNBC |
| FDA accelerated approval in April 2021 for mUC |
| Belantamab mafodotin |
Humanised IgG1 |
BCMA |
Protease-
resistant maleimidohexanoic linker |
MMAF |
Relapsed or refractory multiple myeloma in adults who have received at least four prior therapies |
FDA accelerated approval in 2020 |
| Loncastuximab tesirine- lpyl |
Humanised IgG1 |
CD19 |
Valine–alanine dipeptide |
PDB dimer |
Relapsed or refractory large B-cell lymphoma |
FDA accelerated approval in April 2021 |
| Tisotumab vedotin |
IgG1 |
Tissue factor (TF) |
mc–val–cit–PABC |
MMAE |
Recurrent or metastatic cervical cancer in patients with disease progression during or following chemotherapy |
FDA accelerated approval in September 2021 |
| Moxetumomab Pasudotox |
- |
CD22 |
Recombinant covalently fused |
Pseudotox |
Relapsed or refractory HCL who received at least two prior systemic therapies |
FDA approval in September 2018 |
This entry is adapted from the peer-reviewed paper 10.3390/ddc2020020