SARS-CoV-2 infection, discovered and isolated in Wuhan City, Hubei Province, China, causes acute atypical respiratory symptoms and has led to profound changes in lives. COVID-19 is characterized by a wide range of complications, which include pulmonary embolism, thromboembolism and arterial clot formation, arrhythmias, cardiomyopathy, multiorgan failure, and more. The disease has caused a worldwide pandemic, and despite various measures such as social distancing, various preventive strategies, and therapeutic approaches, and the creation of vaccines, the novel coronavirus infection (COVID-19) still hides many mysteries for the scientific community.
- COVID-19
- ROS
- RNS
- endothelial damage
- mitochondrial dysfunction
- Oxidative stress
- Thrombosis
- DIC
1. Endothelial Injury and Thrombosis
2. Oxidative Stress and Endothelial Dysfunction in Liver Sinusoidal Endothelial Cells
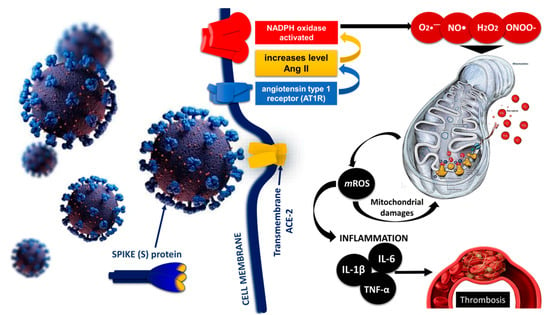
3. Endothelial Damage of the Vascular Layer as a Result of Oxidative Stress in COVID-19
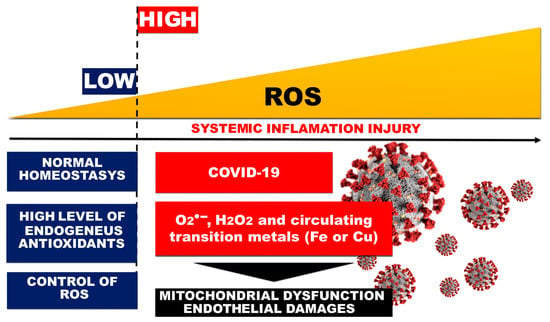
4. DIC and Thrombosis in COVID-19 Cases
This entry is adapted from the peer-reviewed paper 10.3390/ijms241914876
References
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a cellular hub in infection and inflammation. Int. J. Mol. Sci. 2021, 22, 11338.
- Li, D.; Ding, X.; Xie, M.; Tian, D.; Xia, L. COVID-19-associated liver injury: From bedside to bench. J. Gastroenterol. 2021, 56, 218–230.
- Zapotoczny, B.; Braet, F.; Kus, E.; Ginda-Mäkelä, K.; Klejevskaja, B.; Campagna, R.; Chlopicki, S.; Szymonski, M. Actin-spectrin scaffold supports open fenestrae in liver sinusoidal endothelial cells. Traffic 2019, 20, 932–942.
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells 2020, 9, 929.
- Romano, C.; Cozzolino, D.; Nevola, R.; Abitabile, M.; Carusone, C.; Cinone, F.; Cuomo, G.; Nappo, F.; Sellitto, A.; Umano, G.R.; et al. Liver Involvement during SARS-CoV-2 Infection Is Associated with a Worse Respiratory Outcome in COVID-19 Patients. Viruses 2023, 15, 1904.
- Akkiz, H. Unraveling the Molecular and Cellular Pathogenesis of COVID-19-Associated Liver Injury. Viruses 2023, 15, 1287.
- Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653.
- Santoro, L.; Zaccone, V.; Falsetti, L.; Ruggieri, V.; Danese, M.; Miro, C.; Di Giorgio, A.; Nesci, A.; D’Alessandro, A.; Moroncini, G.; et al. Role of Endothelium in Cardiovascular Sequelae of Long COVID. Biomedicines 2023, 11, 2239.
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411.
- Vassiliou, A.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793.
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Jankauskas, S.S.; Kansakar, U.; Sardu, C.; Varzideh, F.; Avvisato, R.; Wang, X.; Matarese, A.; Marfella, R.; Ziosi, M.; Gambardella, J.; et al. COVID-19 Causes Ferroptosis and Oxidative Stress in Human Endothelial Cells. Antioxidants 2023, 12, 326.
- Montiel, V.; Lobysheva, I.; Gérard, L.; Vermeersch, M.; Perez-Morga, D.; Castelein, T.; Mesland, J.B.; Hantson, P.; Collienne, C.; Gruson, D.; et al. Oxidative stress-induced endothelial dysfunction and decreased vascular nitric oxide in COVID-19 patients. EBioMedicine 2022, 77, 103893.
- Amponsah-Offeh, M.; Diaba-Nuhoho, P.; Speier, S.; Morawietz, H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants 2023, 12, 281.
- Xu, S.; Ilyas, I.; Weng, J. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709.
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7.
- Fernández-Ayala, D.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147.
- Fragoso-Morales, L.G.; Correa-Basurto, J.; Rosales-Hernández, M.C. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models. Antioxidants 2021, 10, 218.
- Froldi, G.; Dorigo, P. Endothelial dysfunction in Coronavirus disease 2019 (COVID-19): Gender and age influences. Med. Hypotheses 2020, 144, 110015.
- Morris, G.; Bortolasci, C.; Puri, B.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166.
- Miggiolaro, A.F.R.S.; da Silva, F.P.G.; Wiedmer, D.B.; Godoy, T.M.; Borges, N.H.; Piper, G.W.; Oricil, A.G.G.; Klein, C.K.; Hlatchuk, E.C.; Dagostini, J.C.H.; et al. COVID-19 and Pulmonary Angiogenesis: The Possible Role of Hypoxia and Hyperinflammation in the Overexpression of Proteins Involved in Alveolar Vascular Dysfunction. Viruses 2023, 15, 706.
- Giordo, R.; Paliogiannis, P.; Mangoni, A.; Pintus, G. SARS-CoV-2 and endothelial cell interaction in COVID-19: Molecular perspectives. Vasc. Biol. 2021, 3, R15–R23.
- De Pablo-Moreno, J.A.; Serrano, L.; Revuelta, L.J.; Sánchez, M.J.; Liras, A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int. J. Mol. Sci. 2022, 23, 8283.
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205.
- Angelini, D.E.; Kaatz, S.; Rosovsky, R.P.; Zon, R.L.; Pillai, S.; Robertson, W.E.; Elavalakanar, P.; Patell, R.; Khorana, A. COVID-19 and venous thromboembolism: A narrative review. Res. Pract. Thromb. Haemost. 2022, 6, e12666.
- Sutanto, H.; Soegiarto, G. Risk of Thrombosis during and after a SARS-CoV-2 Infection: Pathogenesis, Diagnostic Approach, and Management. Hematol. Rep. 2023, 15, 225–243.
- Niculae, C.; Hristea, A.; Moroti, R. Mechanisms of COVID-19 Associated Pulmonary Thrombosis: A Narrative Review. Biomedicines 2023, 11, 929.
- Maruhashi, T.; Higashi, Y. Pathophysiological Association of Endothelial Dysfunction with Fatal Outcome in COVID-19. Int. J. Mol. Sci. 2021, 22, 5131.
- Gross, P.L.; Chan, N.C. Thromboembolism in Older Adults. Front. Med. 2021, 7, 470016.
- Yamada, S.; Asakura, H. Therapeutic strategies for disseminated intravascular coagulation associated with aortic aneurysm. Int. J. Mol. Sci. 2022, 23, 1296.
- Levi, M.; Iba, T. COVID-19 coagulopathy: Is it disseminated intravascular coagulation? Intern. Emerg. Med. 2021, 16, 309–312.
- Zhou, X.; Cheng, Z.; Luo, L.; Zhu, Y.; Lin, W.; Ming, Z.; Chen, W.; Hu, Y. Incidence and impact of disseminated intravascular coagulation in COVID-19 a systematic review and meta-analysis. Thromb. Res. 2021, 201, 23–29.
- Papageorgiou, C.; Jourdi, G.; Adjambri, E.; Walborn, A.; Patel, P.; Fareed, J.; Elalamy, I.; Hoppensteadt, D.; Gerotziafas, G.T. Disseminated Intravascular Coagulation: An Update on Pathogenesis, Diagnosis, and Therapeutic Strategies. Clin. Appl. Thromb. Hemost. 2018, 9, 8S–28S.
- Connors, J.; Levy, J. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040.
- Singh, P.; Schwartz, R.A. Disseminated intravascular coagulation: A devastating systemic disorder of special concern with COVID-19. Dermatol. Ther. 2020, 33, 14053.
- Page, E.M.; Ariëns, R.A.S. Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb. Res. 2021, 200, 1–8.
- Milovanovic, L.; Hessey, E.; Sebastianski, M.; Keto-Lambert, D.; Vandermeer, B.; Bagshaw, S.M.; Rewa, O. Epidemiology, clinical characteristics and treatment of critically ill patients with COVID-19): A protocol for a living systematic review. BMJ Open 2021, 11, e042008.
- Chang, R.; Mamun, A.; Dominic, A.; Le, N.T. SARS-CoV-2 Mediated Endothelial Dysfunction: The Potential Role of Chronic Oxidative Stress. Front. Physiol. 2021, 11, 605908.
- Pincemail, J.; Cavalier, E.; Charlier, C.; Cheramy–Bien, J.-P.; Brevers, E.; Courtois, A.; Fadeur, M.; Meziane, S.; Goff, C.L.; Misset, B.; et al. Oxidative Stress Status in COVID-19 Patients Hospitalized in Intensive Care Unit for Severe Pneumonia. A Pilot Study. Antioxidants 2021, 10, 257.
- Soetedjo, N.N.M.; Iryaningrum, M.R.; Damara, F.A.; Permadhi, I.; Sutanto, L.B.; Hartono, H.; Rasyid, H. Prognostic properties of hypoalbuminemia in COVID-19 patients: A systematic review and diagnostic meta-analysis. Clin. Nutr. ESPEN 2021, 45, 120–126.
- Lambadiari, V.; Korakas, E.; Oikonomou, E.; Bletsa, E.; Kountouri, A.; Goliopoulou, A.; Ikonomidis, I.; Siasos, G. COVID-19, Endothelium and the Cardiometabolic Patient: A Possible Role for Capillary Leak Syndrome. Biomedicines 2022, 10, 2379.
- Behzadifard, M.; Soleimani, M. NETosis and SARS-COV-2 infection related thrombosis: A narrative review. Thromb. J. 2022, 20, 13.