Cyclin-dependent kinase (CDK) 4 and its functional homolog CDK6 are two structurally related kinases with biochemical and biological similarities. Despite having few differences in some of their activities, these enzymes are constantly expressed throughout the cell cycle and, with their partners, D-cyclins, are fundamental for integrating mitogenic and antimitogenic extracellular signals, among which stimulating factors, cytokines, cell–cell contacts and other factors are included, representing a boundary between the environment and the cell cycle machinery. The cyclin D-CDK4/6 complex is a driving force that controls the transition from the G1 to the S phases. Also, the INK4 (the cyclin D-CDK4/6 inhibitor molecule) retinoblastoma protein (pRb) pathway regulates cellular proliferation by controlling the G1 to the S cell cycle checkpoint. The dysregulation of this pathway is frequently observed in cancer and contributes to cell cycle progression and persistent growth. CDK4/6 mediates the transition from the G1 phase to the S phase by associating with D-type cyclins and regulating the phosphorylation state of pRb. Unphosphorylated pRb binds and represses the functions of the E2 family (E2F) transcription factors; upon phosphorylation, pRb dissociates from the E2F transcription factors, freeing them to be able to participate in DNA replication and cell division.
- CDK 4/6 inhibitors
- cell-cycle-specific and non-specific resistance
1. Cell-Cycle-Specific Mechanisms
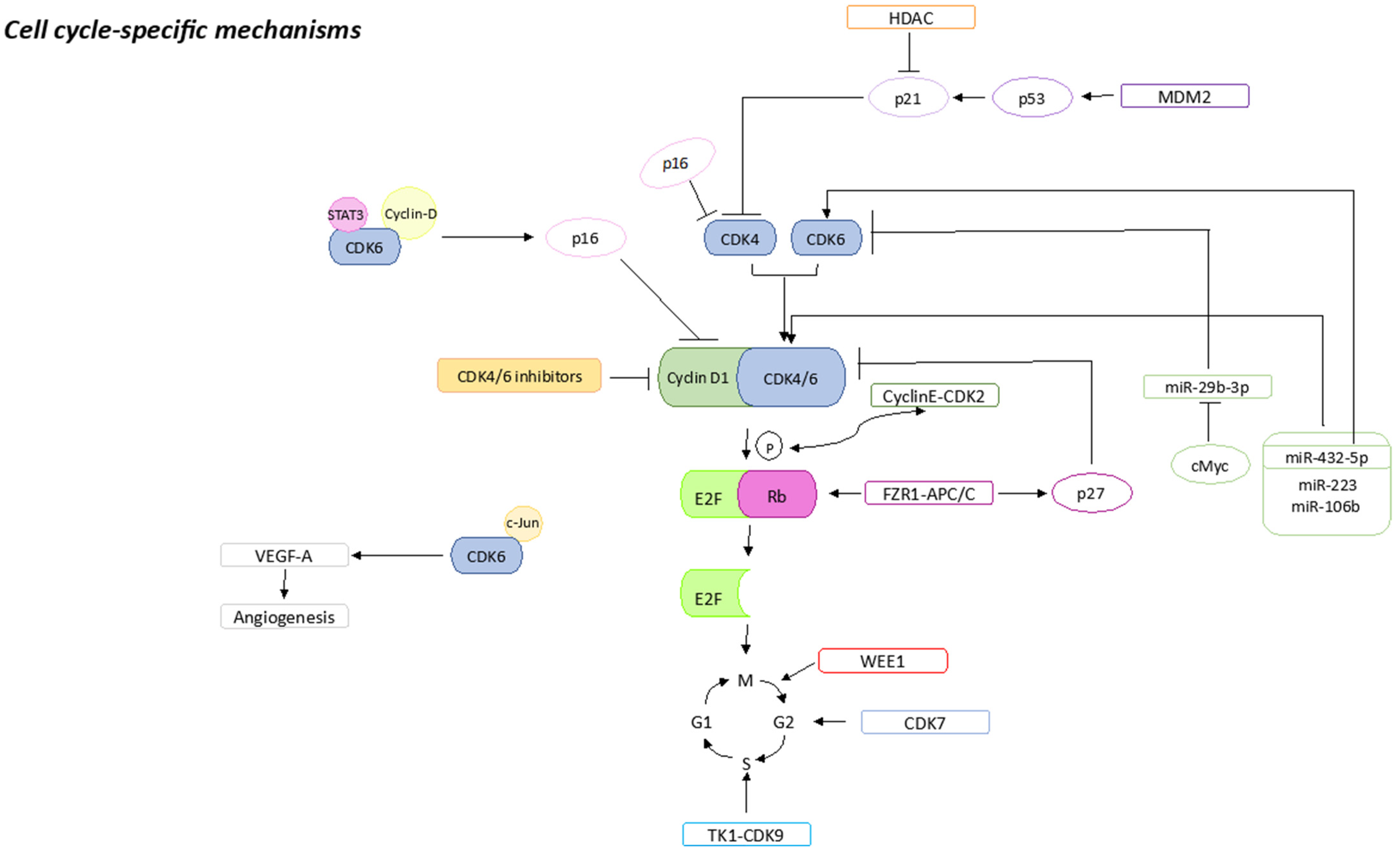
1.1. pRb Loss or Mutations
1.2. p16 Amplification
1.3. CDK2 Amplification
1.4. E2F Amplification
1.5. CDK7 Overexpression
1.6. CDK6 Amplification
1.7. WEE1 Overexpression
1.8. MDM2 Overexpression
1.9. HDACs Activation
1.10. FZR1 Loss
1.11. TK1 Activation
1.12. miRNAs Expression
1.13. S6K1 Amplification
2. Cell-Cycle-Non-Specific Mechanisms
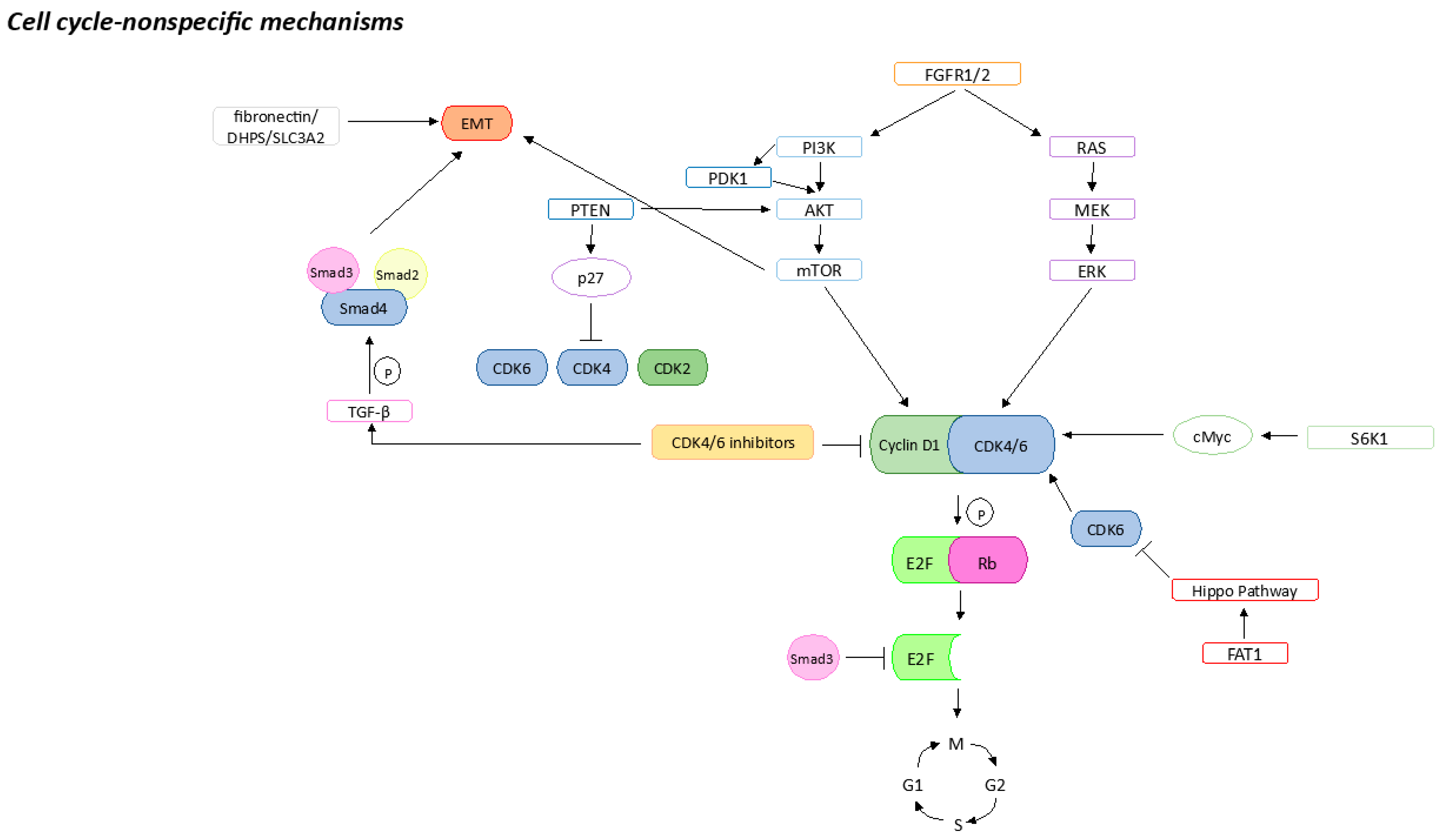
2.1. FGFR Pathway Activation
2.2. PI3K/AKT/mTOR Pathway Activation
2.3. MAPK Pathway Activation
2.4. Hippo Pathway Inhibition by FAT1
2.5. Epithelial–Mesenchymal Transition
2.6. Apoptosis Failure
2.7. Stemness Properties
This entry is adapted from the peer-reviewed paper 10.3390/ijms241914427
References
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504.
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430.
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol. 2015, 16, 25–35.
- Huang, J.; Zheng, L.; Sun, Z.; Li, J. CDK4/6 Inhibitor Resistance Mechanisms and Treatment Strategies (Review). Int. J. Mol. Med. 2022, 50, 128.
- Shapiro, G.I.; Edwards, C.D.; Kobzik, L.; Godleski, J.; Richards, W.; Sugarbaker, D.J.; Rollins, B.J. Reciprocal Rb Inactivation and P16INK4 Expression in Primary Lung Cancers and Cell Lines. Cancer Res. 1995, 55, 505–509.
- Lukas, J.; Parry, D.; Aagaard, L.; Mann, D.J.; Bartkova, J.; Strauss, M.; Peters, G.; Bartek, J. Retinoblastoma-Protein-Dependent Cell-Cycle Inhibition by the Tumour Suppressor P16. Nature 1995, 375, 503–506.
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle 2012, 11, 2756–2761.
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Brasó-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; et al. High P16 Expression and Heterozygous RB1 Loss Are Biomarkers for CDK4/6 Inhibitor Resistance in ER+ Breast Cancer. Nat. Commun. 2022, 13, 5258.
- Gladden, A.B.; Diehl, J.A. Cell Cycle Progression without Cyclin E/CDK2: Breaking down the Walls of Dogma. Cancer Cell 2003, 4, 160–162.
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 Inhibitors Is Associated with Selection of Polyploid Cells in CCNE1-Amplified Ovarian Cancer. Clin. Cancer Res. 2013, 19, 5960–5971.
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313.
- Jin, X.; Ge, L.P.; Li, D.Q.; Shao, Z.M.; Di, G.H.; Xu, X.E.; Jiang, Y.Z. LncRNA TROJAN Promotes Proliferation and Resistance to CDK4/6 Inhibitor via CDK2 Transcriptional Activation in ER+ Breast Cancer. Mol. Cancer 2020, 19, 87.
- Diehl, J.A. Cycling to Cancer with Cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231.
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol. Cell 2013, 50, 250–260.
- Martin, L.-A.; Pancholi, S.; Ribas, R.; Gao, Q.; Simigdala, N.; Nikitorowicz-Buniak, J.; Johnston, S.; Dowsett, M. Abstract P3-03-09: Resistance to Palbociclib Depends on Multiple Targetable Mechanisms Highlighting the Potential of Drug Holidays and Drug Switching to Improve Therapeutic Outcome. Cancer Res. 2017, 77 (Suppl. S4).
- Tigan, A.S.; Bellutti, F.; Kollmann, K.; Tebb, G.; Sexl, V. CDK6-a Review of the Past and a Glimpse into the Future: From Cell-Cycle Control to Transcriptional Regulation. Oncogene 2016, 35, 3083–3091.
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.I.; et al. A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 2016, 30, 359–360.
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881.
- Efeyan, A.; Ortega-Molina, A.; Velasco-Miguel, S.; Herranz, D.; Vassilev, L.T.; Serrano, M. Induction of P53-Dependent Senescence by the MDM2 Antagonist Nutlin-3a in Mouse Cells of Fibroblast Origin. Cancer Res. 2007, 67, 7350–7357.
- Cox, L.S. Multiple Pathways Control Cell Growth and Transformation: Overlapping and Independent Activities of P53 and P21Cip1/Waf1/Sdi1. J. Pathol 1997, 183, 134–140.
- Portman, N.; Chen, J.; Lim, E. MDM2 as a Rational Target for Intervention in CDK4/6 Inhibitor Resistant, Hormone Receptor Positive Breast Cancer. Front. Oncol. 2021, 11, 777867.
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903.
- Dash, B.C.; El-Deiry, W.S. Phosphorylation of P21 in G2/M Promotes Cyclin B-Cdc2 Kinase Activity. Mol. Cell. Biol. 2005, 25, 3364–3387.
- Zhou, Y.; Jin, X.; Ma, J.; Ding, D.; Huang, Z.; Sheng, H.; Yan, Y.; Pan, Y.; Wei, T.; Wang, L.; et al. HDAC5 Loss Impairs RB Repression of Pro-Oncogenic Genes and Confers CDK4/6 Inhibitor Resistance in Cancer. Cancer Res. 2021, 81, 1486–1499.
- Ramanujan, A.; Tiwari, S. APC/C and Retinoblastoma Interaction: Cross-Talk of Retinoblastoma Protein with the Ubiquitin Proteasome Pathway. Biosci. Rep. 2016, 36, e00377.
- The, I.; Ruijtenberg, S.; Bouchet, B.P.; Cristobal, A.; Prinsen, M.B.W.; Van Mourik, T.; Koreth, J.; Xu, H.; Heck, A.J.R.; Akhmanova, A.; et al. Rb and FZR1/Cdh1 Determine CDK4/6-Cyclin D Requirement in C. Elegans and Human Cancer Cells. Nat. Commun. 2015, 6, 5906.
- Fujita, T.; Liu, W.; Doihara, H.; Wan, Y. Regulation of Skp2-P27 Axis by the Cdh1/Anaphase-Promoting Complex Pathway in Colorectal Tumorigenesis. Am. J. Pathol. 2008, 173, 217–228.
- He, Q.; Zou, L.; Zhang, P.A.; Lui, J.X.; Skog, S.; Fornander, T. The Clinical Significance of Thymidine Kinase 1 Measurement in Serum of Breast Cancer Patients Using Anti-TK1 Antibody. Int. J. Biol. Markers 2000, 15, 139–146.
- Bjöhle, J.; Bergqvist, J.; Gronowitz, J.S.; Johansson, H.; Carlsson, L.; Einbeigi, Z.; Linderholm, B.; Loman, N.; Malmberg, M.; Söderberg, M.; et al. Serum Thymidine Kinase Activity Compared with CA 15-3 in Locally Advanced and Metastatic Breast Cancer within a Randomized Trial. Breast Cancer Res. Treat. 2013, 139, 751–758.
- Del Re, M.; Bertolini, I.; Crucitta, S.; Fontanelli, L.; Rofi, E.; De Angelis, C.; Diodati, L.; Cavallero, D.; Gianfilippo, G.; Salvadori, B.; et al. Overexpression of TK1 and CDK9 in Plasma-Derived Exosomes Is Associated with Clinical Resistance to CDK4/6 Inhibitors in Metastatic Breast Cancer Patients. Breast Cancer Res. Treat. 2019, 178, 57–62.
- Lin, S.L.; Chang, D.C.; Ying, S.Y.; Leu, D.; Wu, D.T.S. MicroRNA MiR-302 Inhibits the Tumorigenecity of Human Pluripotent Stem Cells by Coordinate Suppression of the CDK2 and CDK4/6 Cell Cycle Pathways. Cancer Res. 2010, 70, 9473–9482.
- Pulito, C.; Cristaudo, A.; La Porta, C.; Zapperi, S.; Blandino, G.; Morrone, A.; Strano, S. Oral Mucositis: The Hidden Side of Cancer Therapy. J. Exp. Clin. Cancer Res. 2020, 39, 210.
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667–2680.e7.
- Ji, W.; Zhang, W.; Wang, X.; Shi, Y.; Yang, F.; Xie, H.; Zhou, W.; Wang, S.; Guan, X. C-Myc Regulates the Sensitivity of Breast Cancer Cells to Palbociclib via c-Myc/MiR-29b-3p/CDK6 Axis. Cell Death Dis. 2020, 11, 760.
- Fingar, D.C.; Blenis, J. Target of Rapamycin (TOR): An Integrator of Nutrient and Growth Factor Signals and Coordinator of Cell Growth and Cell Cycle Progression. Oncogene 2004, 23, 3151–3171.
- Mo, H.; Liu, X.; Xue, Y.; Chen, H.; Guo, S.; Li, Z.; Wang, S.; Li, C.; Han, J.; Fu, M.; et al. S6K1 Amplification Confers Innate Resistance to CDK4/6 Inhibitors through Activating c-Myc Pathway in Patients with Estrogen Receptor-Positive Breast Cancer. Mol. Cancer 2022, 21, 171.
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129.
- Luqmani, Y.A.; Graham, M.; Coombes, R.C. Expression of Basic Fibroblast Growth Factor, FGFR1 and FGFR2 in Normal and Malignant Human Breast, and Comparison with Other Normal Tissues. Br. J. Cancer 1992, 66, 273–280.
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 2010, 70, 2085–2094.
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293.
- Mao, P.; Kusiel, J.; Cohen, O.; Wagle, N. Abstract PD4-01: The Role of FGF/FGFR Axis in Resistance to SERDs and CDK4/6 Inhibitors in ER+ Breast Cancer. Cancer Res. 2018, 78 (Suppl. S4), PD4-01.
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499.
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of MTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-Term Growth Inhibition in Estrogen Receptor-Positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920.
- Costa, C.; Ye, W.; Ly, A.; Hosono, Y.; Murchi, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85.
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B.; et al. INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov. 2022, 12, 356–371.
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 2019, 10, 1373.
- Raimondi, L.; Raimondi, F.M.; Pietranera, M.; Di Rocco, A.; Di Benedetto, L.; Miele, E.; Lazzeroni, R.; Cimino, G.; Spinelli, G.P. Assessment of Resistance Mechanisms and Clinical Implications in Patients with KRAS Mutated-Metastatic Breast Cancer and Resistance to CDK4/6 Inhibitors. Cancers 2021, 13, 1928.
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo Pathway in Cancer, Fibrosis, Wound Healing and Regenerative Medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494.
- Finn, R.S.; Crown, J.P.; Ettl, J.; Schmidt, M.; Bondarenko, I.M.; Lang, I.; Pinter, T.; Boer, K.; Patel, R.; Randolph, S.; et al. Efficacy and Safety of Palbociclib in Combination with Letrozole as First-Line Treatment of ER-Positive, HER2-Negative, Advanced Breast Cancer: Expanded Analyses of Subgroups from the Randomized Pivotal Trial PALOMA-1/TRIO-18. Breast Cancer Res. 2016, 18, 67.
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8.
- Bae, S.N.; Arand, G.; Azzam, H.; Pavasant, P.; Torri, J.; Frandsen, T.L.; Thompson, E.W. Molecular and Cellular Analysis of Basement Membrane Invasion by Human Breast Cancer Cells in Matrigel-Based in Vitro Assays. Breast Cancer Res. Treat. 1993, 24, 241–255.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Zelivianski, S.; Cooley, A.; Kall, R.; Jeruss, J.S. Cyclin-Dependent Kinase 4-Mediated Phosphorylation Inhibits Smad3 Activity in Cyclin D-Overexpressing Breast Cancer Cells. Mol. Cancer Res. 2010, 8, 1375–1387.
- Yang, J.; Song, K.; Krebs, T.L.; Jackson, M.W.; Danielpour, D. Rb/E2F4 and Smad2/3 Link Survivin to TGF-Beta-Induced Apoptosis and Tumor Progression. Oncogene 2008, 27, 5326–5338.
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965.
- Geller, C.; Maddela, J.; Tuplano, R.; Runa, F.; Adamian, Y.; Güth, R.; Soto, G.O.; Tomaneng, L.; Cantor, J.; Kelber, J.A. Fibronectin, DHPS and SLC3A2 Signaling Cooperate to Control Tumor Spheroid Growth, Subcellular EIF5A1/2 Distribution and CDK4/6 Inhibitor Resistance. bioRxiv 2023.
- Johnston, S.; Puhalla, S.; Wheatley, D.; Ring, A.; Barry, P.; Holcombe, C.; Boileau, J.F.; Provencher, L.; Robidoux, A.; Rimawi, M.; et al. Randomized Phase II Study Evaluating Palbociclib in Addition to Letrozole as Neoadjuvant Therapy in Estrogen Receptor-Positive Early Breast Cancer: PALLET Trial. J. Clin. Oncol. 2019, 37, 178–189.
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 4120–4134.
- Zeng, X.; Liu, C.; Yao, J.; Wan, H.; Wan, G.; Li, Y.; Chen, N. Breast Cancer Stem Cells, Heterogeneity, Targeting Therapies and Therapeutic Implications. Pharmacol. Res. 2021, 163, 105320.
- Wang, S.; Bei, Y.; Tian, Q.; He, J.; Wang, R.; Wang, Q.; Sun, L.; Ke, J.; Xie, C.; Shen, P. PFKFB4 Facilitates Palbociclib Resistance in Oestrogen Receptor-Positive Breast Cancer by Enhancing Stemness. Cell Prolif. 2023, 56, e13337.