1. Introduction
The insulin-IGF system is formed by insulin, two insulin-like growth factors (IGF-I and IGF-II), four cell-membrane receptors (insulin receptor-A (IR-A), insulin receptor-B (IR-B), insulin-like growth factor-I receptor (IGF-IR) and insulin-like growth factor receptor-II (IGF-II-R)) and six IGF-binding proteins (IGFBP-1-6), several IGFBP- related proteins and IGFBP proteases [
1,
2,
3,
4]. All IGFBPs can bind both IGF-I and IGF-II (however with different binding affinity for some) [
5]. Only the unbound forms of IGFs are thought to interact with the IGF-IR and the IGF-II receptor [
6].
The IGF-I gene comprises a highly conserved sequence and contains six exons, which give rise to heterogeneous mRNA transcripts by a combination of multiple transcription initiation sites and alternative splicing [
7]. These multiple transcripts code in humans for different precursor IGF-I polypeptides, namely the IGF-IEa, IGF-IEb and IGF-IEc isoforms, which also undergo posttranslational modifications, such as proteolytic processing and glycosylation [
7]. Differential biological activities have been reported for the different IGF-I isoforms and thus both common and unique or complementary pathways exist for the IGF-I isoforms to promote biological effects [
7].
As IGFs and insulin as well as the IGF-IR and the IRs share high sequence homology, they are able to bind and activate each other’s cognate receptors but with considerably lower avidity. The IGF-IR can bind IGF-I and IGF-II with equally high affinity (10
−10 M) whereas its affinity for insulin (10
−8 M) is much lower [
8]. In the past it was thought that the IGFs and the IGF-IR predominantly mediated growth-promoting effects whereas insulin and the IRs predominantly mediated metabolic effects [
9,
10]. However, in certain circumstances IGF-I and insulin can mediate very similar responses [
11]. Nevertheless, IGF-I and IGF-II play a crucial factor in the regulation of growth, proliferation, differentiation, migration and survival of cells. In addition, activation of IGF-IR and its intracellular pathways has been found to be essential for growth of cancers [
12].
IGF-IIR regulates the amount of circulating and tissue IGF-II by transporting IGF-II into the cell and degrading it [
13]. IGF-II can also bind to the IGF-IR with high affinity [
13].
2. IGF-I and the IGF-I Receptor
The IGF-IR is displayed on the cell surface and expressed by nearly all human tissues and cell types [
5,
9,
17]. Surface density of the IGF-IR represents an important determinant of the magnitude of responses to IGF-I and the signaling pattern it provokes [
18]. The IGF-IR is a heterotetrameric transmembrane protein composed of two alpha and two beta subunits which are linked by disulfide bonds [
19,
20]. The beta subunit of the IGF-IR consists of a short extracellular domain which is involved in linkage to the alpha subunits, a transmembrane domain and a cytoplasmic domain containing tyrosine kinase activity [
21]. The beta subunit contains a consensus ATP-binding sequence and multiple tyrosine residues that are phosphorylated following ligand binding to the alpha subunit [
21]. Binding of IGF-I or another ligand to the alpha subunit of the IGF-IR, induces a closer proximity of regions within the transmembrane domain resulting in autophosphorylation of three intracellular tyrosine residues (Tyr
1131, Tyr
1135, and Tyr
1136) within the beta subunit [
21,
22,
23].
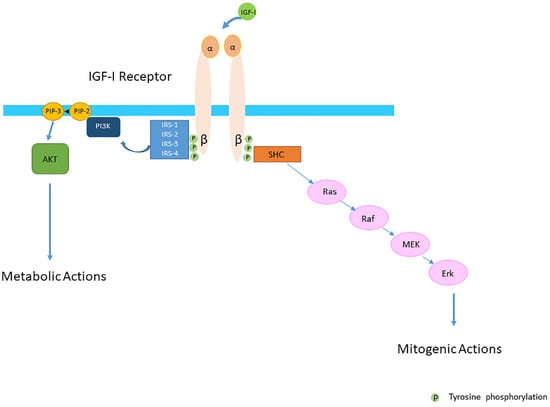
The conventional view was that the IGF-IR was exclusively a tyrosine kinase receptor and that the binding of IGF-I to the IGF-IR was essential to start the intracellular downstream signal cascade (
Figure 1). In this model, the activated receptor recruited and phosphorylated intracellularly substrates as the insulin receptor substrate proteins (IRSs) and SH2 containing collagen-related proteins (SHC) (
Figure 1). Tyrosine phosphorylation of the IRSs in turn activated then the phosphatidylinositol 3-kinase (PI3K-Akt) pathway and its various biological responses, while tyrosine phosphorylation of SHC induced downstream signaling activation through the Ras/Raf/MEK/Erk pathway [
24,
25] (
Figure 1).
Figure 1. The Insulin-like Growth Factor-I (IGF-IR) is a transmembrane protein composed of two alpha (α) and two beta (β) subunits. The conventional view was that the IGF-IR was exclusively a tyrosine kinase receptor and that the binding of IGF-I to the IGF-IR started the intracellular downstream signal cascade. In this model IGF-I or IGF-II binding to the IGF-IR promotes tyrosine kinase activity and autophosporylation of the beta subunit of the IGF-IR. Intracellularly the activated IGF-IR receptor recruits phosphorylated substrates Insulin receptor substrates (IRSs) and SH2 containing collagen-related proteins (SHC). Tyrosine phosphorylation of IRSs and SHC proteins induces downstream signaling activation through the PI3K-AKT and Ras/Raf/MEK/Erk pathways. It was further thought that activation of the PI3K-AKT pathway had predominantly metabolic effects whereas activation of the Ras/Raf/MEK/Erk pathway had predominantly mitogenic effects.
3. The IGF-IR and Endocytosis
Many signaling receptors internalize via clathrin-coated pits [
26]. Endocytosis of signaling receptors is widely recognized to confer control on cellular signaling responsiveness [
27]. Ligand-induced activation typically increases receptor endocytic rate, and internalized receptors engage molecular sorting machineries that specify subsequent transport via divergent lysosomal and recycling routes [
27]. These events, in turn, determine the degree to which cellular ligand responsiveness is attenuated (“down-regulated”) or sustained (“re-sensitized”) under conditions of prolonged or repeated ligand exposure [
27].
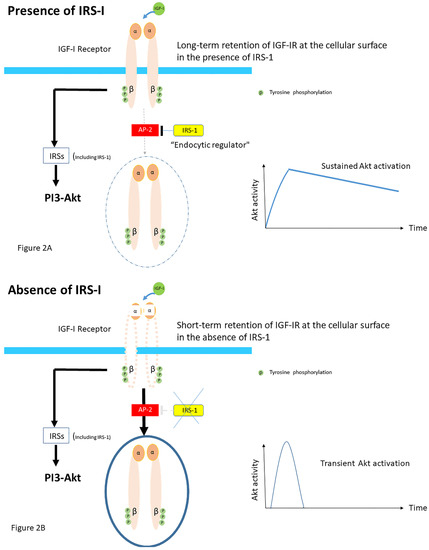
The molecular basis for the close interactions between IGF-IR endocytosis and its signaling components is still poorly understood. Recently, it has been suggested that the ability of insulin receptor substrate-1 (IRS-1) to interact with the clathrin adapter protein AP2, which is essential for endocytosis, plays an important role in IGF-IR internalization [
28]. Overexpression of IRS-I resulted in the accumulation of activated IGF-IR at the cellular membrane [
28]. Conversely, knockdown of IRS-I induced faster internalization of IGF-IRs [
28]. These data suggest that IRS-1 inhibits the recruitment of IGF-IR into clathrin-coated structures; the ability of IRS-I to bind to AP-2 avoids rapid endocytosis of the IGF-IR and prolongs its activity at the cell surface in HEK293T cells [
28] (
Figure 2A). In contrast, accelerating IGF-IR endocytosis via IRS-1 depletion induces the shift from sustained to transient Akt signaling [
28] (
Figure 2B). Thus, independent of its classic role as an adaptor in IGF-I receptor signaling, IRS-1 has a role as an endocytic regulator of IGF-I receptor that ensures sustained IGF bioactivity, while IRS-1 degradation could be a trigger to internalize the IGF-IR [
29].
Figure 2. Proposed role of Insulin receptor substrate-1 (IRS-1). IRS-1 modulates how long ligand-activated IGF-IR remains at the cell surface before undergoing endocytosis in mammalian cells. IRS-1 interacts with the clathrin adaptor complex AP2. (A) In the presence of the IRS-1/AP2-complex in the cell IGF-IR endocytosis after the ligand stimulation is delayed. Mechanistically, IRS-1 inhibits the recruitment of IGF-IR into clathrin-coated structures; for this reason, IGF-IR avoids rapid endocytosis and prolongs its activity on the cell surface and this results in sustained activation of the AKT pathway. (B) In absence of IRS-1/AP2- complex in the cell, there is only short-term retention of the IGF-IR at the cell surface and IGF-IR endocytosis is accelerated. This results in a transient activation of the AKT pathway (Modified from Yoneyama et al. IRS-1 acts as an endocytic regulator of IGF-I receptor to facilitate sustained IGF signaling. eLIFE, 2018; 7. pii: e32893).
For the IGF-IR, ubiquitination also increases upon ligand binding [
30]. The IGF-IR has been demonstrated to be a substrate for three ubiquitin ligases: Mdm2, (in human malignant melanoma cells), c-Cbl (HEK293 cells and human osteosarcoma cell lines U2OS and SAOS2) and Nedd4 (in mouse embryo fibroblasts). [
30,
31]. Mdm2 was originally described to control IGF-IR ubiquitination and thereby causing its degradation by the proteasome system [
32]. Subsequently β-arrestins, known to be involved in the regulation of G-protein-coupled receptors (GPCRs), have also been identified as adaptor proteins to bring the oncoprotein Mdm2 to the IGF-IR in mouse embryo fibroblasts [
33,
34]. In addition, while removing the IGF-IR from the cell surface and inhibiting the “classical” kinase signaling pathway, β-arrestins may redirect the signaling wave through ERK [
33] Ubiquitination may thus induce receptor internalization and degradation, but also enhance IGF-IR signaling [
35] (which will be further addressed in the paragraph “the complexity of the post-receptor IGF-IR/IR pathways” below).
4. Structural Differences and Overlap between the IGF-IR and the IRs
It is hypothesized that the IGF-IR and IRs are created by gene duplication of common precursor receptor molecule [
36]. Due to structural and functional homology, IGF-I and insulin can bind to (and activate) both IGF-IR and the IRs, as discussed above [
37]. IGF-IR and IRs show 48% amino acid sequence homology [
20]. Structural differences between the beta-subunit and kinase domains of the IGF-IR and the IRs leading to differences in substrate interactions may be (partly) responsible for IGF-I and insulin specificity as has been found in various cell types (rat-1 fibroblasts, murine skin keratinocytes and in NIH-3T3-fibroblasts) [
38]. However, the signal transduction by the receptors may not be limited to its activation at the cell surface [
39].
In addition to signaling through the classical tyrosine kinase pathways, it has been found that the IGF-IRs and IRs (in cells derived from C57Bl/6 mice) can emit signals in the unoccupied state through some yet-to-be-defined non-canonical pathways [
40]. Boucher et al. demonstrated that cells lacking the IGF-IR and IR exhibit a major decrease in expression of multiple imprinted genes and microRNAs [
40].
Although the IGF-IR and IRs have both distinct and overlapping functions, it has been suggested that in vivo specificity of the IGFs and insulin are at least in part reflected by the timing of the expression of the IGF-IR and IRs in target tissues in combination with ligand concentration and availability [
41]. It has been further suggested that IGF-I and the IRs act as identical portals for the regulation of gene expression and that the differences between IGF-I and insulin effects are due to a modulation of the amplitude of the signal created by the specific ligand receptor interaction [
41].
5. The IGF-IR and the IRs May Form Hybrids in the Human Body
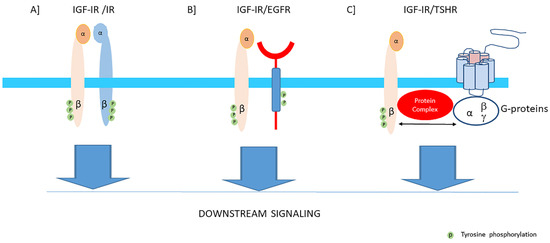
In cells and tissues where both significant levels of the IGF-IRs and IRs are present, hybrids may be formed consisting of an alpha-beta subunit of the IGF-IR linked by disulfide bonds to an alpha-beta subunit of the IR [
42] (
Figure 3A). They are formed in the endoplasmic reticulum before they reach the cell surface [
43]. Two splice variants of the hybrid receptors exist for the IR because the IR is expressed (as above discussed) either with (IR-B) or without 12 amino acids encoded by exon 11 (IR-A) [
44]. Thus both IR-A/IGF-IR (Hybrid A) and IR-B/IGF-IR (Hybrid B) receptors can be formed. Although the biological functions of these hybrid receptors is still unclear, it has been suggested that hybrid receptors may play a role in the overlapping functions of IGF-I and insulin [
21]. Several studies (in baby hamster kidney cells, NIH3T3 cells overexpressing IGFR and CHO cells overexpressing IR-B) have suggested that cells by increasing the relative expression level of the IGF-IR above that of the IR lose their insulin sensitivity because hybrid receptors bind insulin with low affinity [
45,
46]. In addition, binding of insulin to the alpha-beta subunit of the IR which is part of a hybrid, may result in autophosphorylation of its ow beta subunit and, following transphosphorylation of the beta subunit of the IGF-IR, may result in a signal for growth, [
9]. In contrast, when IGF-I binds to the alpha-beta subunit of the IGF-IR, this may activate the beta subunit of the IRs by the same mechanisms and thereby activate growth (IR-A) or metabolism (IR-B) [
9]. Although this latter mechanism could explain why hybrids may stimulate metabolic functions when stimulated by IGF-I, most functional studies have found that hybrid receptors behave more like IGF-IRs than IRs [
47]. It has been hypothesized that this prioritization of hybrid receptors to IGF-I results from the ability of IGF-I to activate monomeric IGF-IR whereas, in contrast, dimerization of the IR has been considered necessary to induce a response to insulin [
45].
Figure 3. The IGF-I receptor may form hybrids with the insulin receptor, many other tyrosine kinase receptors outside the insulin-IGF system and G-protein coupled receptors. The figure shows three examples: (A) Hybrids may be formed consisting of an alpha-beta subunit of the IGF-IR linked by disulfide bonds to an alpha-beta subunit of the IR. Downstream signaling of both receptors converge via the canonical PI3K-Akt and ERK signaling pathways. Most functional studies have found that hybrid receptors behave more like IGF-IRs than IRs (See also text). (B) Hybrids may be formed consisting of an alpha-beta subunit of the IGF-IR linked and a monomer of the epidermal growth factor receptor (EGFR) which is also a tyrosine kinase receptor. Downstream signaling of both receptors converge via the canonical PI3K-Akt and ERK signaling pathways. Therefore, inhibition of one receptor of these hybrids may shift the signaling pathway in favor of the other available counterpart receptor. (C) The Thyroid Stimulating Hormone Receptor (TSH receptor), a typical G-protein coupled receptor, may form functional hybrids with the IGF-IR in the cellular membrane by forming a common protein complex. Bidirectional crosstalk between the IGF-IR and TSHR has been demonstrated. Stimulation of the IGF-IR by IGF-I/IGF-IR agonists may trigger the classical signaling pathway of the IGF-IR, leading to downstream kinase-cascade signaling activation. In addition, stimulation of the IGF-IR by IGF-I/IGF-IR agonists may also utilize components of G-protein coupled receptor (GPCR) signaling and activate pathways conventionally used by TSHR.
In cells and tissues where both significant levels of the IGF-IRs and IRs are present, hybrids may be formed consisting of an alpha-beta subunit of the IGF-IR linked by disulfide bonds to an alpha-beta subunit of the IR [
42] (
Figure 3A). They are formed in the endoplasmic reticulum before they reach the cell surface [
43]. Two splice variants of the hybrid receptors exist for the IR because the IR is expressed (as above discussed) either with (IR-B) or without 12 amino acids encoded by exon 11 (IR-A) [
44]. Thus both IR-A/IGF-IR (Hybrid A) and IR-B/IGF-IR (Hybrid B) receptors can be formed. Although the biological functions of these hybrid receptors is still unclear, it has been suggested that hybrid receptors may play a role in the overlapping functions of IGF-I and insulin [
21]. Several studies (in baby hamster kidney cells, NIH3T3 cells overexpressing IGFR and CHO cells overexpressing IR-B) have suggested that cells by increasing the relative expression level of the IGF-IR above that of the IR lose their insulin sensitivity because hybrid receptors bind insulin with low affinity [
45,
46]. In addition, binding of insulin to the alpha-beta subunit of the IR which is part of a hybrid, may result in autophosphorylation of its ow beta subunit and, following transphosphorylation of the beta subunit of the IGF-IR, may result in a signal for growth, [
9]. In contrast, when IGF-I binds to the alpha-beta subunit of the IGF-IR, this may activate the beta subunit of the IRs by the same mechanisms and thereby activate growth (IR-A) or metabolism (IR-B) [
9]. Although this latter mechanism could explain why hybrids may stimulate metabolic functions when stimulated by IGF-I, most functional studies have found that hybrid receptors behave more like IGF-IRs than IRs [
47]. It has been hypothesized that this prioritization of hybrid receptors to IGF-I results from the ability of IGF-I to activate monomeric IGF-IR whereas, in contrast, dimerization of the IR has been considered necessary to induce a response to insulin [
45].
It has been further hypothesized that IGF-IR/IR hybrids may affect tumor biology [
48]. Specific downregulation of the IGF-IR by agents solely targeting the IGF-IR diminishes hybrid formation and this thereby enhances holo-IR formation [
48]. An enhanced holo-IR formation results in an increase of insulin sensitivity [
48]. As the IR, especially the IR-A, may also activate (post-receptor) signaling pathways involved in growth similar to the IGF-IR, the development of agents simultaneous targeting both IR-A and IGF-IR may be necessary to disrupt the malignant phenotype of cancers cells that are influenced by actions of the insulin-IGF system [
48].
The IGF-IR may also heterodimerize with receptor tyrosine kinases (RTKs) outside the insulin-IGF system [
49]. Heterodimerization of the IGF-IR with the EGFR is well-established [
50] (
Figure 3B). Downstream signaling of both receptors converge via the canonical PI3K-Akt and ERK signaling pathways. Therefore inhibition of one receptor in these hybrids may shift the signaling pathway in favor of the other available counterpart receptor [
49,
51,
52]. Thus the compensatory signaling may be bidirectional [
53]. Moreover, evidence exists that IGF-IR can activate independently downstream EGFR pathways and this may subsequently result in EFGR tyrosine kinase inhibitor (TKI) resistance [
52]. The IGF-IR signaling pathway shows also cross-talk with the growth hormone receptor (GHR), thyroid stimulating hormone receptor (TSHR) (
Figure 3C), estrogen receptor (ER), androgen receptor (AR) and human epidermal growth factor receptor 2 (HER-2) signaling pathways [
54,
55,
56,
57,
58,
59].
Thus, it has become clear in recent years that the IGF-IR signaling pathway is far more complex than previously thought. It contains many points of regulation and shows signal divergence and cross-talk with many other signaling pathways at the receptor and post-receptor level. However, a big challenge for the (near) future will be how all this new knowledge about the IGF-IR signaling pathways can be translated into clinical practice and improve diagnosis and treatment of diseases.
This entry is adapted from the peer-reviewed paper 10.3390/cells9040862