Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Material innovation plays a very important role in technological progress and industrial development. Traditional experimental exploration and numerical simulation often require considerable time and resources. A new approach is urgently needed to accelerate the discovery and exploration of new materials. Machine learning can greatly reduce computational costs, shorten the development cycle, and improve computational accuracy.
- machine learning
- material screening
- property prediction
- material synthesis
1. Prediction of Material Properties
Machine learning (ML) has gained prominence in recent years in predicting material properties due to its advantages of high generalization ability and fast computational speed. It has been successfully applied to predict the structure, adsorption, electrical, catalytic, energy storage, and thermodynamic properties of materials. The prediction results could even reach the same accuracy as high-fidelity models with low computational costs.
1.1. Molecular Properties
In the past, it was very time consuming to predict molecular properties based on high-throughput density generalization calculations. ML allows fast and accurate prediction of the structure or properties of molecules, compounds, and materials. In materials science, solubility factors, such as Hansen and Hildebrand solubility, are critical parameters for characterizing the physical properties of various substances. Kurotani et al. [1] successfully developed a solubility prediction model with a unique ML method, the so-called in-phase DNN (ip-DNN). This algorithm started with the analysis of input data (including NMR information, refractive index, and density). The solubility was then speculated in a multi-step approach by predicting intermediate elements, such as molecular components and molecular descriptors. An intermediate regression model was also utilized to improve the accuracy of the prediction. A website dedicated to the established solubility prediction methods has also been developed, which is available free of charge. Liang et al. [2] proposed a generalized ML method based on ANNs to predict polymer compatibility (the total miscibility of polymers with each other at the molecular scale). The authors built a database by collecting data from scattered literature through natural language processing techniques. By using the proposed method, predictions could be made based on the basic molecular structure of the blended polymers and the blended compositions (as an auxiliary). This generalized approach yielded some results in illustrating polymer compatibility. A prediction accuracy of no less than 75% was achieved on a dataset containing 1400 entries in their model. Zeng et al. [3] developed an atomic table CNN that could predict the band gap and ground energy. The model accuracy exceeded that of standard DFT calculations. Furthermore, this model could accurately predict superconducting transition temperatures and distinguish between superconductors and non-superconductors. With the help of this model, 20 potential superconductor compounds with high superconducting transition temperatures were screened out.
1.2. Band Gap
The band gap size not only determines the energy band structure of a material but also affects its electronic structure and optical properties. Recently, researchers have applied ML to forecast the band gap of various materials. Venkatraman [4] developed an algorithm for band gap prediction based on a rule-based ML framework. With descriptors derived from elemental compositions, this model accurately and quickly predicted the band gap of various materials. After testing on two independent sets, this model obtained squared correlations > 0.85, with errors smaller than those of most density generalization calculations, improving the material screening performance. Xu et al. [5] developed an ML model called support vector regression (SVR) for predicting the band gaps of polymers. They used training data obtained from DFT computations and generated descriptors using Dragon software. After feature selection, the SVR model using 16 key features achieved high accuracy in predicting polymer band gaps. The SVR model with a Gaussian kernel function performed the best, with a determination coefficient (R2) of 0.824 and a root mean square error (RMSE) of 0.485 in leave-one-out cross-validation. The authors also provided correlation analysis and sensitivity analysis to understand the relationship between the selected features and the band gaps of polymers. Several polymer samples with targeted band gaps were designed based on the analysis and validated through DFT calculations and model predictions. Espinosa et al. [6] proposed a vision-based system to predict the electronic band gaps of organic molecules using deep learning techniques. The system employed a multichannel 2D CNN and a 3D CNN to recognize and classify 2D projected images of molecular structures. The training and testing datasets used in the research were derived from the Organic Materials Database (OMDB-GAP1). The results showed that the proposed CNN model achieved a mean absolute error of 0.6780 eV and an RMSE of 0.7673 eV, outperforming other ML methods based on conventional DFT. These findings demonstrate the potential of CNN models in materials science applications using orthogonal image projections of molecules. Wang et al. [7] explored the use of ML techniques to accurately predict the band gaps of semiconductor materials. The authors applied a stacking approach, which combined the outputs of multiple baseline models, to enhance the performance of band gap regression. The effectiveness of different models was tested using a benchmark dataset and a newly established complex database. The results showed that the stacking model had the highest R2 value in both datasets, indicating its superior performance. The improvement percentages of various evaluation metrics for the stacking model compared to other baseline models range from 3.06% to 33.33%. Overall, the research demonstrated the excellent performance of the stacking approach in band gap regression. On the basis of generalized gradient approximation (GGA) band gap information of crystal structures and materials, Na et al. [8] established an ML method that used the tupleswise graph neural network (TGNN) algorithm for the accurate band gap prediction of crystalline compounds. The TGNN algorithm showed strong superiority in predicting the band gap of four different open databases. It has better accuracy for 48,835 samples of G0W0 (a widely used technique in which the self-energy is expressed as the convolution of a noninteracting Green’s function (G0) and a screened Coulomb interaction (W0) in the frequency domain) band gaps than the standard density generalized theory without high computational costs. Moreover, this model could be extended to project other valuable properties.
1.3. Energy Storage Performance
Energy storage is a key step in determining the efficiency, stability, and reliability of power supply systems [9]. Exploring the energy storage performance of materials is critical to energy storage, and ML accelerates the exploration process. Feng et al. [10] collected over one thousand composite energy storage performance data points from the open literature and utilized ML to analyze and build a predictive model. The prediction accuracies of the RF, SVM, and neural network were 84.1%, 80.9%, and 70.6%, respectively. They then added processed visual information data of the composite into the dataset, resulting in improved prediction accuracies of 91.9%, 68.9%, and 81.6% for the three models, respectively. This demonstrated that the dispersion of the filler in the matrix is an important factor affecting the maximum energy storage density of the composite. The authors also analyzed the weights of each descriptor in the RF model and explored the effects of various parameters on the energy storage of the material. Figure 1 shows the logic diagram of their ML models. Yue et al. [11] utilized the packing dielectric constant, packing size, and packing content as descriptors to predict the energy storage density of polymer matrix composites. High-throughput random breakdown simulations were performed on 504 datasets. The simulation results were then applied as an ML database and combined with classical dielectric prediction equations. They experimentally validated the predictions, including the dielectric constant and breakdown strength. This work provides insights into the design and fabrication of polymer matrix composites with enhanced energy density for applications in capacitive energy storage. Ojin et al. [12] built four traditional ML models and two graph neural network models. Through them, 32,026 heat capacity structures were predicted using a high-precision deep graph attention network. Additionally, the correlation between heat capacity and structure descriptors was inspected. A total of 22 structures were predicted to have high heat capacity, and the results were further validated by DFT analysis. Through the combination of ML and minimal DFT queries, this study provides a path to accelerating the discovery of new thermal energy storage materials.
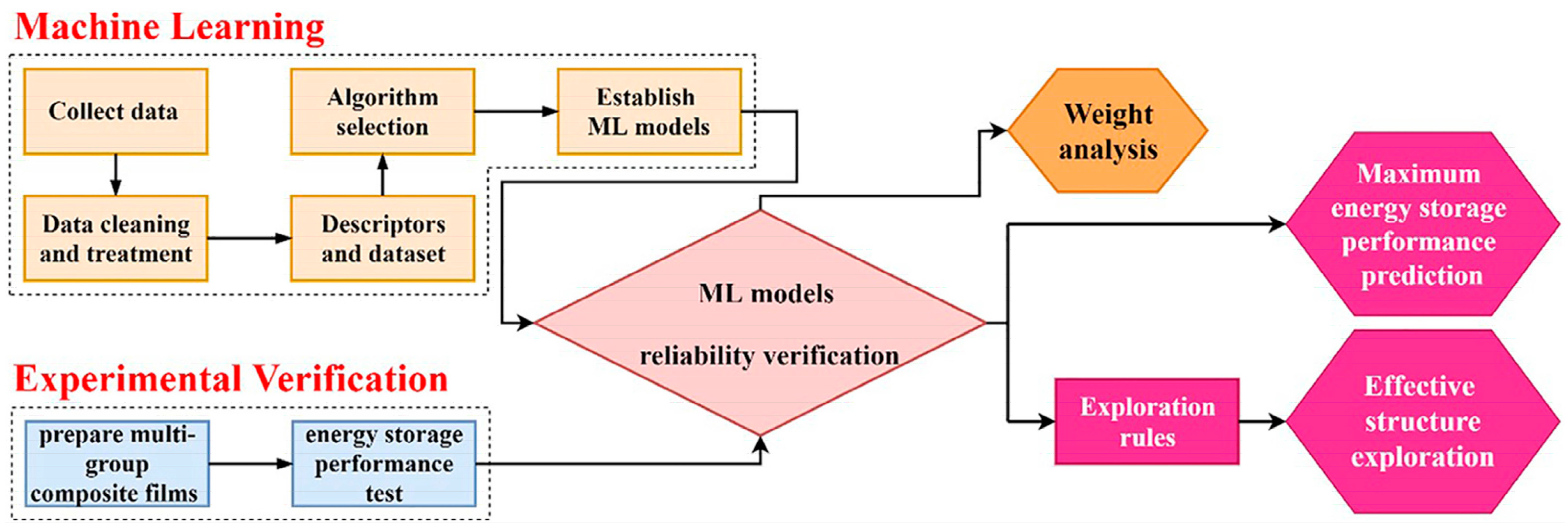
Figure 1. Logic diagram of predicting the maximum energy density and exploring the potential effective structure of composites through the ML method, reproduced with permission from [10].
1.4. Structural Health
Structural health monitoring (SHM) utilizes engineering, scientific, and foundational knowledge to prevent damage to property and life. The core of the field of construction informatics is the transmission, processing, and visualization of architectural information, providing effective methods for monitoring structural changes [13][14]. ML provides effective methods for monitoring structural changes. Dang et al. [15] proposed a cloud-based digital twin framework for SHM employing deep learning. The framework consists of physical components, device measurements, and digital models formed by combining different sub-models including mathematical, finite element, and ML sub-models. The data interactions among the physical structure, digital model, and human interventions were enhanced by using cloud computing infrastructure and a user-friendly web application. The feasibility of the framework was demonstrated through case studies of the damage detection of model bridges and real bridge structures utilizing deep learning algorithms, with a high accuracy of 92%. Dong et al. [16] discussed the use of the eXtreme gradient boosting (XGBoost) algorithm for predicting concrete electrical resistivity in SHM (Figure 2a). The proposed XGBoost-algorithm-based prediction model considers all potential influencing factors simultaneously. A database of 800 experimental instances was used to train and test the model. The results showed that the XGBoost model achieved satisfactory predictive performance. The study also identified the importance of curing age and cement content in electrical resistivity measurement results. The XGBoost algorithm was chosen for its high performance, ease of use, and better prediction accuracy than other algorithms. The bond effect between the reinforcement and concrete guarantees the combined action of the two materials. This is a critical factor that affects the mechanical properties of reinforced concrete components and structures, e.g., bearing capacity and ductility [17]. Gao et al. [18] developed a new solution for evaluating the bond strength of an FRP using AI-based models. Two hybrid models, the imperialist competitive algorithm (ICA)-ANN and the artificial bee colony (ABC)-ANN, were designed and compared. The results showed that the ICA-ANN model had a higher predictive ability than the ABC-ANN model. The proposed hybrid models can be used as a suitable substitute for empirical models in evaluating FRP bond strength in concrete samples. Li et al. [19] utilized ML approaches to estimate the bond strength between ultra-high-performance concrete (UHPC) and reinforcing bars. A new database was created by integrating data from multiple published works. Nine ML models, including linear models, tree models, and ANNs, were implemented to train bond strength estimators based on the database. The results showed that the ANN and RF models achieved the highest estimation performances, surpassing empirical formulas. The study also analyzed the relative importance of different factors in determining bond strength. Overall, the research provides a data-driven approach to estimating bond strength and contributes to the understanding of bond performance between UHPC and reinforcing bars. Su et al. [20] applied three ML approaches (multiple linear regression, SVM, and ANN) to predict the interfacial bond strength between FRPs and concrete (Figure 2b). They trained these models using two datasets containing experimental results from single-lap shear tests, employed random search and grid search to find the optimal hyperparameters, and analyzed input variables’ contributions using partial dependence plots. They also developed a stacking strategy to improve prediction accuracy. The results showed that the SVM approach had the best accuracy and efficiency. They concluded that ML methods are feasible and efficient for predicting the bond strength of FRP laminates in reinforced concrete structures.

1.5. Nanomaterial Toxicity
It has been proven that ML can be used to identify nanomaterial properties and exposure conditions that influence cellular and organism toxicity, thus providing information required for risk assessment and safe-by-design approaches in the development of new nanomaterials [21]. Huang et al. [22] combined ML with high-throughput in vitro bioassays to develop a model to predict the toxicity of metal oxide nanoparticles to immune cells, as shown in Figure 3. In the training, test, and experimental validation sets, the ML model displayed prediction accuracies of 97%, 96%, and 91%, respectively. ML methods were used to identify features that encode information on immune toxicity. These features are crucial for the scientific design of future experiments and for the accurate depiction of nanotoxicity. According to Gousiadoua et al. [23], advanced ML techniques were applied to create nano quantitative structure–activity relationship (QSAR) tools for modeling the toxicity of metallic and metal oxide nanomaterials, both coated and uncoated, with various core compositions tested on embryonic zebrafish at various dosage concentrations. Based on both computed and experimental descriptors, the scientists identified a set of properties most relevant for assessing nanomaterial toxicity and successfully correlated these properties with zebrafish physiological responses. It has been concluded that for the group of metal and metal oxide nanomaterials, the core chemical composition, concentration, and properties are influenced by the nanomaterial surface and medium composition (such as zeta potential and agglomerate size), which have a significant impact on toxicity, even though the ranking of different variables is subject to variation in the analytical method and data model. Generalized nano-QSAR ensemble models offer a promising framework for predicting the toxicity potential of new nanomaterials. Liu et al. [24] presented a meta-analysis of phytosynthesized silver nanoparticles (AgNPs) with heterogeneous features using DTs and RFs. The researchers found that exposure regime (including the time and dose), plant family, and cell type were the most important predictors for cell viability for green AgNPs. In addition, a discussion of the potential effects of major variables (cell assays, inherent nanoparticle properties, and reaction parameters used in biosynthesis) on AgNP-mediated cytotoxicity and model performance was presented to provide a basis for future research. The findings of this study may assist future studies in improving the design of experiments and the development of virtual models or optimizations of green AgNPs for specific applications.
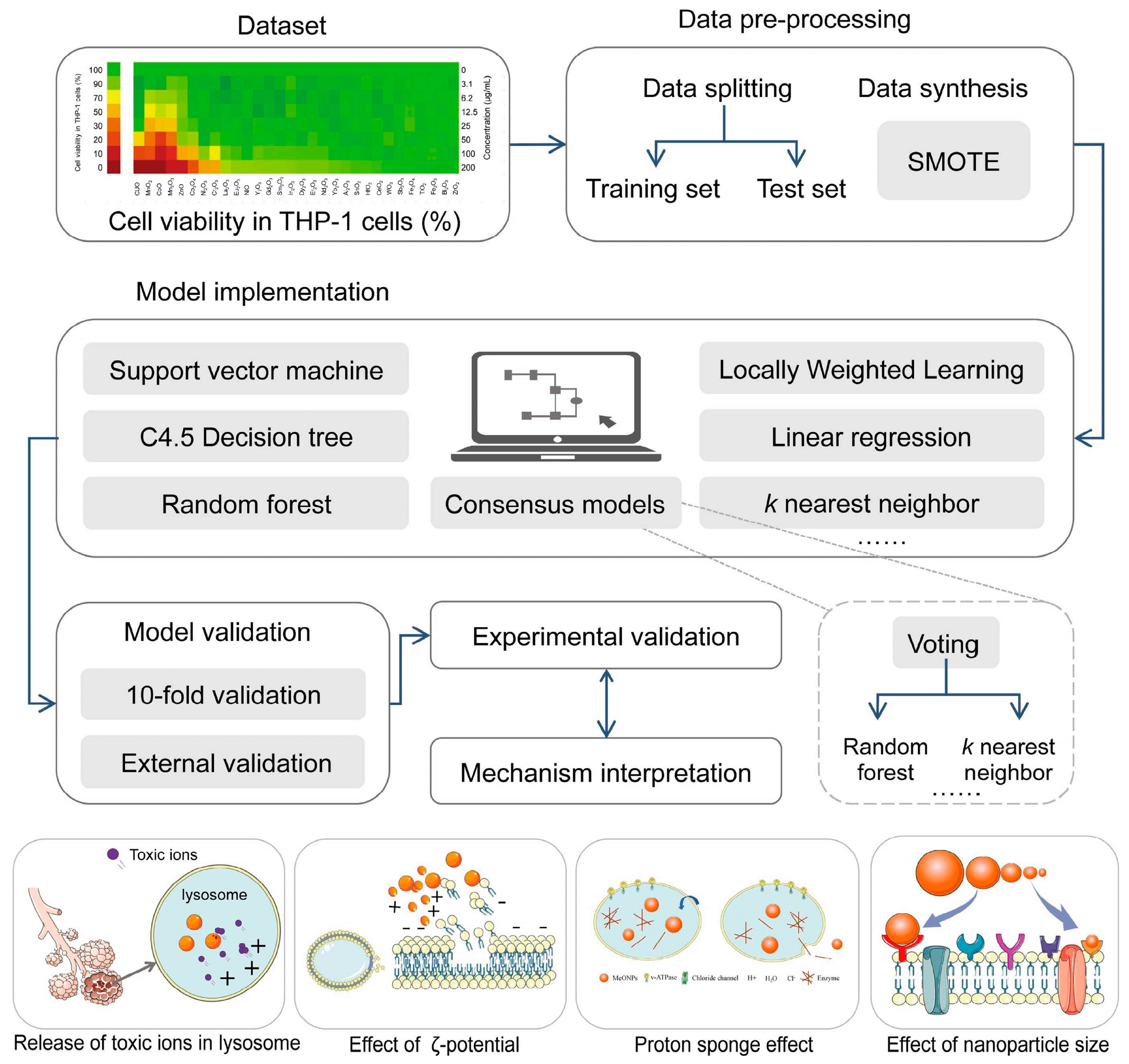
Figure 3. Schematic workflow of data compilation, descriptor generation, machine learning modeling, experimental validation, and mechanism interpretation, reproduced with permission from [22].
1.6. Adsorption Performance of Nanomaterials
Because of their high surface area, ease of functionalization, and affinity toward a wide range of pollutants, nanomaterials are excellent adsorbents [25]. Moosavi et al. [26] applied four machine learning methods to model dye adsorption on 16 activated carbon adsorbents and determined the relationship between adsorption capacity and activated carbon parameters. The results indicated that agro-waste characteristics (pore volume, surface area, pH, and particle size) contributed 50.7% to the adsorption efficiency. Among the agro-waste characteristics, pore volume and surface area were the most important influencing variables, while particle size had a limited impact. With a hypothetical set of approximately 130,000 structures of metal–organic frameworks (MOFs) with methane and carbon dioxide adsorption data at different pressures, Guo et al. [27] established models for estimating gas adsorption capacities using two deep learning algorithms, multilayer perceptrons (MLPs) and long short-term memory (LSTM) networks. The models were evaluated by performing ten iterations of 10-fold cross-validations and 100 holdout validations. The performance of the MLP and LSTM models was similar with high accuracy of prediction. Those models that predicted gas adsorption at a higher pressure performed better than those that predicted gas adsorption at a lower pressure. In particular, deep learning models were more accurate than RF models reported in the literature when predicting gas adsorption capacities at low pressures. Deep learning algorithms were found to be highly effective in generating models capable of accurately predicting the gas adsorption capacities of MOFs.
2. Accelerated Materials Synthesis and Design
In addition to being widely utilized for predicting material properties, ML also plays a pivotal role in the synthesis of new materials. During the past few years, ML has made significant progress in the exploration of novel materials, such as highly efficient molecular organic light-emitting diodes [28], low thermal hysteresis shape memory alloys [29], and piezoelectric materials with large electrical strain [30]. The use of ML for materials synthesis not only significantly speeds up novel material discovery but also provides insight into the basic composition changes in materials from big data.
2.1. Chalcogenide Materials
Chalcogenide materials can be used in a variety of photovoltaic and energy devices, including light-emitting diodes, photodetectors, and batteries. ML has promoted the development of high-performance chalcogenide materials [31]. Li et al. [32] proposed an ML model based on an RF algorithm for speculating the formation of ABX3 and A2B′B″X6 compound chalcogenides. With geometric and electrical parameters, the RF classification model reached 96.55% accuracy for ABX3 samples and 91.83% accuracy for A2B′B″X6 samples. A total of 241 ABX3 chalcogenides with a 95% probability of formation were filtered from 15,999 candidate compounds, and a total of 1131 A2B′B″X6 chalcogenides with a 99% probability of formation were filtered from 417,835 candidate compounds. The method presented in their work could offer valuable enlightenment for the acceleration of discovering perovskites. Liu et al. [33] used data from 397 ABO3 compounds and nine parameters (e.g., tolerance factor and octahedral factor) as input variables for ML. The gradient-enhanced DT obtained by training was compared as the optimal model by 10-fold cross-validation of the average accuracy. A total of 331 chalcogenides were filtered by the model from 891 data points with a classification accuracy of 94.6%. Omprakash et al. [34] compiled a model including organometallic salt chalcogenides to 2D chalcocite and its corresponding band gaps. An ML model for predicting all types of chalcocite band gaps was then trained using a graphical representation learning technique. The model could accurately estimate the band gap within a few milliseconds with an average absolute error of 0.28 eV. Wang et al. [35] applied unsupervised learning to discover quaternary chalcogenide semiconductors (I2-II-IV-X4) and were successful in screening eight of these materials with good photoconversion efficiency despite a data shortage. This method shortens the material screening cycle and facilitates rapid material discovery.
2.2. Catalytic Materials
In traditional experiments, it is difficult to design efficient catalytic materials in a short time because a clear reaction mechanism is required [36]. ML can rapidly extract the relationship between the structure and performance of catalytic materials and effectively expedite the development process of new catalytic materials. Zhang et al. [37] employed a gradient boosting algorithm to build an ML model. The model utilized four key stability and catalytic features of graphene-loaded single-atom catalysts as targets to find catalytic materials suitable for electro-hydrogenation nitrogen reactions. With this model, a total of 45 catalytic materials with efficient catalytic performance were successfully screened from 1626 samples. The model could be operated for the rapid screening of other electrocatalysts. Figure 4 illustrates their computational framework. Wei et al. [38] developed an ML model, which was applied in a Bayesian optimization framework to obtain molybdenum disulfide (MoS2) catalysts with stable hydrogen reaction activity. To explore the structure–property relationship of the samples optimized by the ML technique, nine electrochemical characterizations were performed to verify the results, including SEM, TEM, XRD, and XPS. A strong correlation was found between the structure of the optimized MoS2 and its hydrogen evolution reaction performance. Hueffel et al. [39] reported an unsupervised ML workflow that uses only five experimental data points, which could be used to accelerate the recognition of binuclear palladium (Pd) catalysts. Based on their method, some phosphine ligands were successfully predicted and experimentally verified from 348 ligands, including those that had never been synthesized before, which formed binuclear Pd(I) complexes on Pd(0) and Pd(II) species. Their strategy plays an important role in studying the formation mechanisms of Pd catalyst species, as well as the further integration of ML into catalytic research.
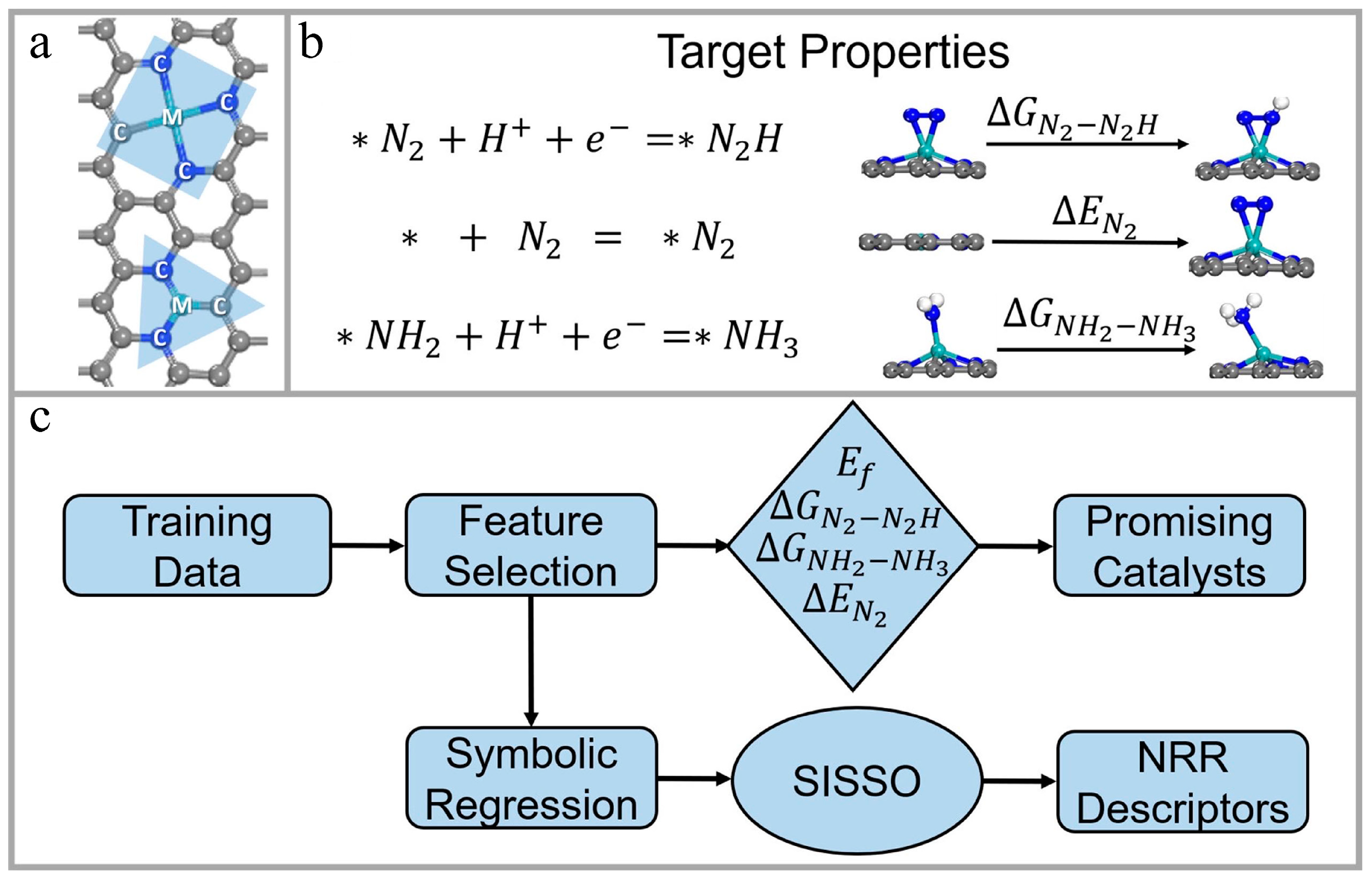
Figure 4. Catalyst structures, target properties, and computational framework. (a) Structural representation of three-coordinated and four-coordinated configurations. Letter “M” represents the central metal atom, and letter “C” represents the coordinating atom of M. (b) Target properties for describing the N2 fixation performance of the catalyst. (c) ML screening and descriptor building framework of their work. Reproduced with permission from [37].
2.3. Superconducting Materials
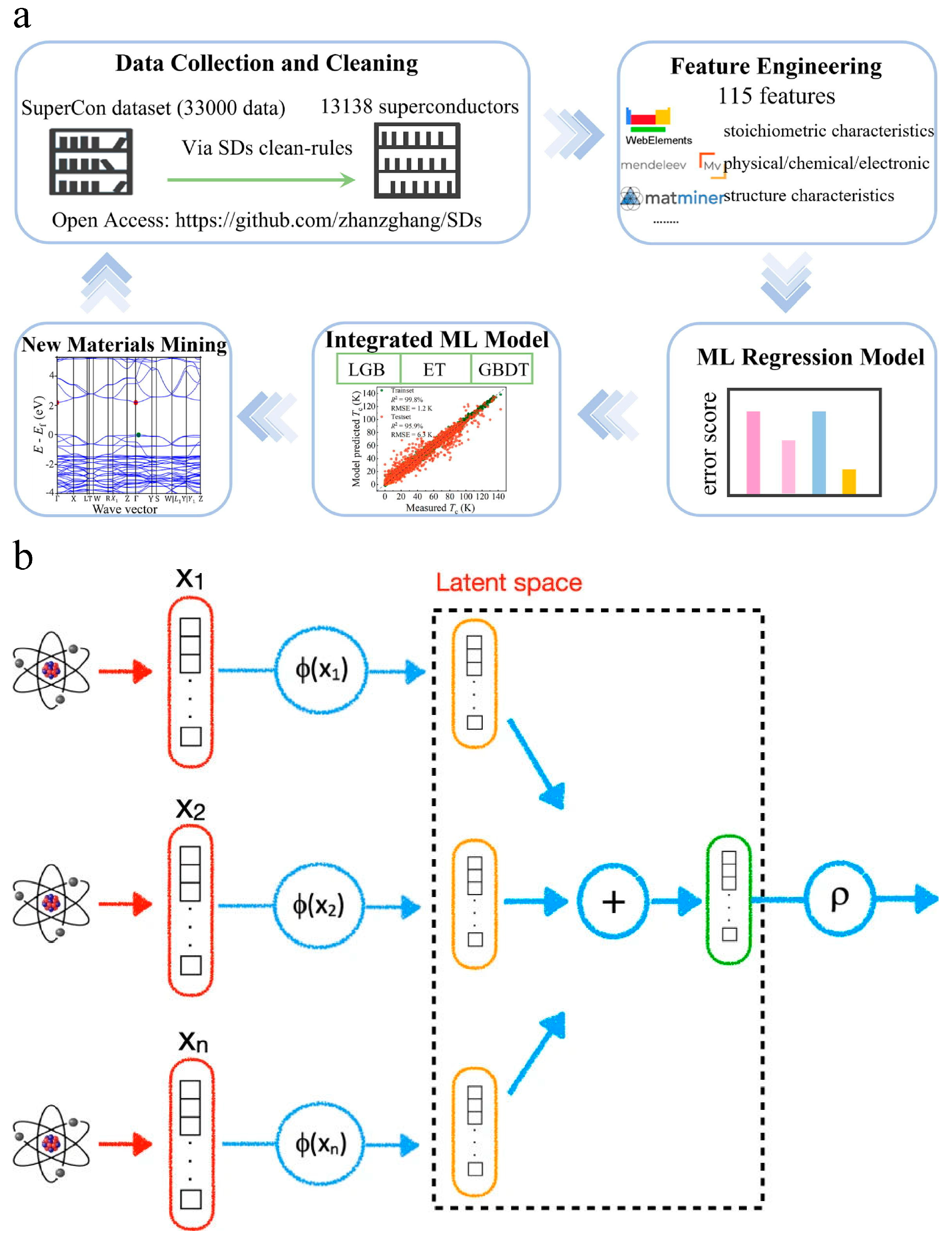
Superconductivity, intrinsically regulated by finite phonon-coupled electron–electron attractions, has aroused decades of intense research interest in condensed matter physics. The development and prediction of upcoming superconducting materials with high critical temperatures are essential in many applications. ML-guided iterative experimentation may outperform standard high-throughput screening for discovering breakthrough materials in high-temperature superconductors [40][41]. Zhang et al. [42] developed an integrated ML model to accurately and robustly predict the critical temperature (Tc) of superconducting materials (Figure 5a). They used open-source materials data, ML models, and data mining methods to explore the correlation between chemical features and Tc values. The integrated model combined three basic algorithms (gradient boosting decision tree, extra tree, and light gradient boosting machine) to improve the prediction accuracy. The model achieved an R2 of 95.9% and an RMSE of 6.3 K. The study also identified the importance of various material features in Tc prediction, with thermal conductivity playing a critical role. The integrated model was used to screen out potential superconducting materials with Tc values beyond 50.0 K. This research provides insights for accelerating the exploration of high-Tc superconductors. Roter et al. [43] used ML to predict new superconductors and their critical temperatures. They constructed a database of superconductors and their chemical compositions and applied this information to train ML models. They achieved an R2 of approximately 0.93, which was comparable to or higher than similar estimates based on other AI techniques. They also discussed factors that limit learning and suggested possible ways to overcome them. The researchers used both unsupervised and supervised ML techniques, including singular value decomposition and KNN, to improve their models’ accuracy. They achieved a classification accuracy of 96.5% and an R2 of approximately 0.93 for predicting critical temperatures. They also employed their models to predict several new superconductors with high critical temperatures. However, the authors noted that incorrect entries in the database can lead to outliers in the predictions. Pereti et al. [44] proposed an ML approach to identify new superconducting materials. They utilized DeepSet technology, which allows them to input the chemical constituents of the compounds without predetermined ordering (Figure 5b). The method was successful in classifying materials as superconducting and quantifying their critical temperature. The trained neural network was then used to search through a mineralogical database for candidates that might be superconducting. Three materials were selected for experimental characterization, and superconductivity was confirmed in two of them. This was the first time a superconducting material was identified using AI methods. The results demonstrated the effectiveness of the DeepSet network in predicting the critical temperatures of superconducting materials.
2.4. Nanomaterial Outcome Prediction
Rapid advancements in materials synthesis techniques have led to more and more attention being paid to nanomaterials, including nanocrystals, nanorods, nanoplates, nanoclusters, and nanocrystalline thin films. Materials of this class offer enhanced physical and chemical tunability across a range of systems, including inorganic semiconductors, metals, and molecular crystals. A nanomaterial is defined as a material with a dimension smaller than 100 nanometers in at least one dimension. Unlike bulk materials, nanomaterials possess different physical and chemical properties due to their unique size and shape. This technology has a broad array of application prospects, including the conversion and storage of energy, the restoration of water, medical treatment, and the storage and processing of data.
Using experimental data, Xie et al. [45] reported the development of an ML-aided method for predicting the crystallization tendency of metal–organic nanocapsules (MONCs). A prediction accuracy of >91% was achieved by using the XGBoost model. Furthermore, they synthesized a set of new crystalline MONCs using the derived features and chemical hypotheses from the XGBoost model. The results of this study demonstrate that ML algorithms can assist chemists in finding the optimal reaction parameters from a large number of experimental parameters more efficiently. Figure 6 shows a schematic representation of the working flow. Pellegrino et al. [46] tuned the TiO2 nanoparticle morphology using hydrothermal treatment. In their work, an experimental design was employed to investigate the influence of relevant process parameters on the synthesis outcome, enabling ML methods to develop predictive models. After validation and training, the models were capable of accurately predicting the synthesis outcome in terms of nanoparticle size, polydispersity, and aspect ratio. They presented a synthesis method that allows the continuous and precise control of nanoparticle morphology. This method affords the possibility to tune the aspect ratio over a large range from 1.4 (perfect truncated bipyramids) to 6 (elongated nanoparticles) and a length from 20 to 140 nm.
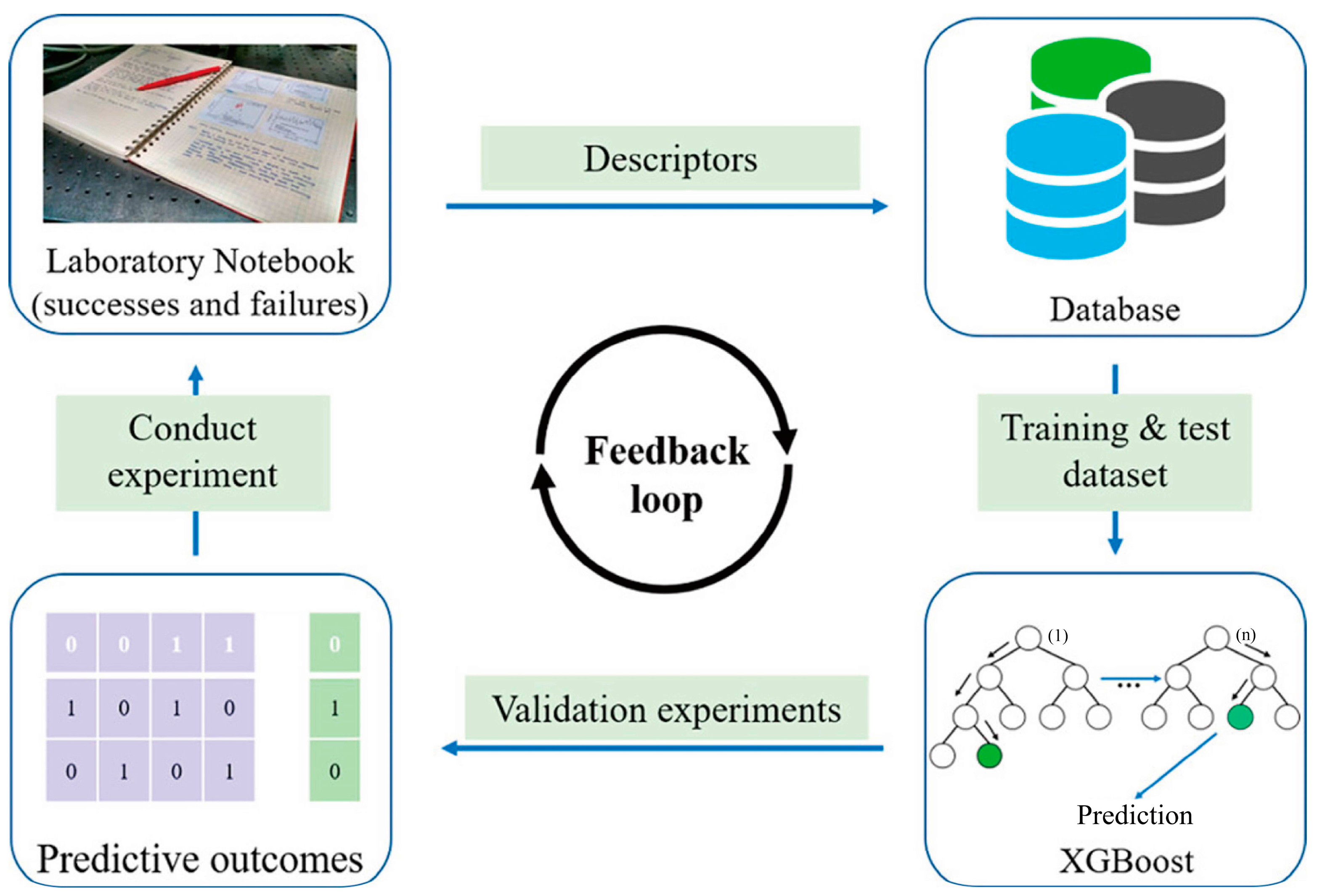
Figure 6. Schematic representation of the working flow when machine learning models are incorporated into the prediction of the crystallization propensity of MONCs, with permission from [45].
2.5. Nanomaterial Synthesis
Nanomaterial synthesis often involves multiple reagents and interdependent experimental conditions. Each experimental variable’s contribution to the final product is generally determined through trial and error, along with intuition and experience. The process of identifying the most efficient recipe and reaction conditions is therefore time consuming, laborious, and resource intensive [47]. In a recent study, Erick et al. [48] used SVM classification and regression models to predict the synthesis of CsPbBr3 nanosheets with controlled layer thicknesses. The SVM classification is shown to accurately predict the likelihood that CsPbBr3 synthesis would form a majority population of quantum-confined nanoplatelets. Additionally, SVM regression can be used to determine the average thickness of the synthesis of CsPbBr3 nanoplatelets with sub-monolayer accuracy. Epps et al. [49] proposed a method that is based on ML experiment selection and high-efficiency autonomous flow chemistry. The approach utilized SVM regression to predict the thickness of the nanoplatelets and was shown to be accurate and reliable. Using this method, inorganic perovskite quantum dots (QDs) in flow were synthesized autonomously. By using less than 210 mL of starting solutions and without user selection, this method synthesized precision tailored QD compositions within 30 h. This would enable the commercialization of these QDs, as well as their integration into various applications. Furthermore, the method could be used for other types of nanomaterials, such as nanorods and nanowires.
2.6. Inverse Design of Nanomaterials
As opposed to the direct approach that leads from the chemical space to the desired properties, inverse design starts with desired properties as the “input” and ends with chemical space as the “output” [50]. In the field of nanomaterials, the complexity of inverse design is enhanced by the finite dimensions and variety of shapes, resulting in a larger design space [51]. The inverse design of nanomaterials was quite challenging in the past. The inverse design of nanomaterials could be explored using interpretable relationships between structure and property generated by ML methods. A new inverse design method for metal nanoparticles based on deep learning was proposed and demonstrated by Wang et al. [52]. In comparison to the least squares method, the calculated results indicated that the inverse design method utilizing the back-propagation network had greater adaptability, a smaller minimum error, and can be adjustable based on S parameters. Inverse design systems based on deep learning neural networks may be applied to the inverse design of nanoparticles of different shapes. In another study, Li et al. [51] demonstrated a novel approach to inverse design using multi-target regression methods using RFs. A multi-target regression model was used with a precursory forward structure–property prediction to capture the most important characteristics of a single nanoparticle before the problem was inverted and a number of structural features were simultaneously predicted. A general workflow has been demonstrated on two nanoparticle datasets, and it has the capacity to predict rapid relationships between properties and structures for guiding further research and development without the need for additional optimization or high-throughput sampling. He et al. [53] employed a DNN to establish mappings between the far-field spectra/near-field distribution and dimensional parameters of three different types of plasmonic nanoparticles, including nanospheres, nanorods, and dimers. Through the DNN, both the forward prediction of far-field optical properties and the inverse prediction of nanoparticle dimensional parameters can be accomplished accurately and efficiently. Figure 7 shows the structure of the reported machine learning model for predicting optical properties and designing nanoparticles.
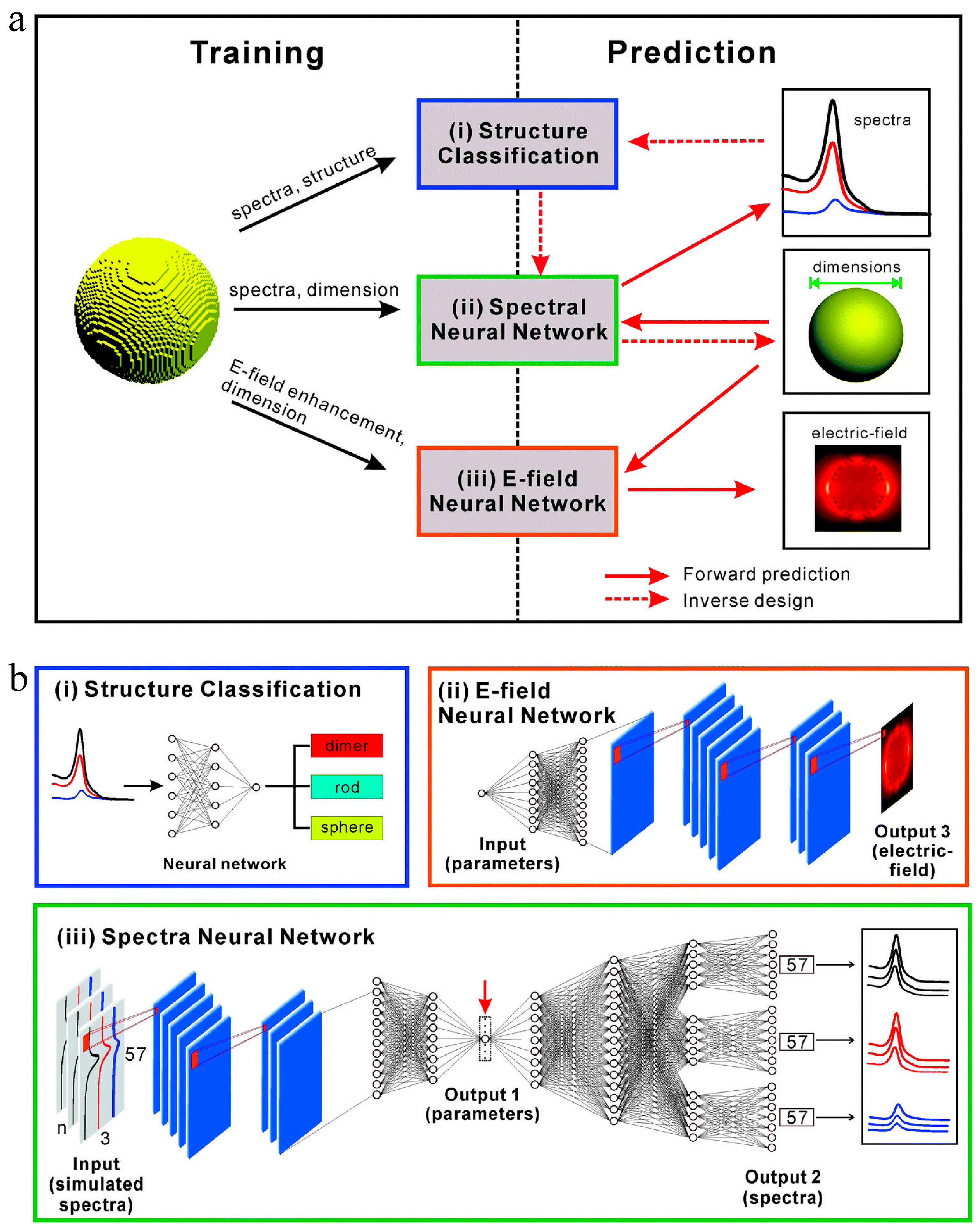
Figure 7. Structures of machine learning models for predicting optical properties and designing nanoparticles. (a) Far- and near-field optical data obtained from the finite-difference time-domain (FDTD) simulations were used to train three different machine learning models: far-field spectra and structural information for (i) structure classification, far-field spectra and dimensions for (ii) the spectral DNN, and near-field enhancement maps and dimensions for (iii) the E-field DNN. After training, machine learning models can be used to perform forward prediction and/or inverse design. The solid and dashed red arrows represent the forward prediction and the inverse design process, respectively. (b) Detailed architecture of the three machine learning models in Figure 1a, with permission from [53].
This entry is adapted from the peer-reviewed paper 10.3390/ma16175977
References
- Kurotani, A.; Kakiuchi, T.; Kikuchi, J. Solubility prediction from molecular properties and analytical data using an in-phase deep neural network (Ip-DNN). ACS Omega 2021, 6, 14278–14287.
- Liang, Z.; Li, Z.; Zhou, S.; Sun, Y.; Yuan, J.; Zhang, C. Machine-learning exploration of polymer compatibility. Cell Rep. Phys. Sci. 2022, 3, 100931.
- Zeng, S.; Zhao, Y.; Li, G.; Wang, R.; Wang, X.; Ni, J. Atom table convolutional neural networks for an accurate prediction of compounds properties. NPJ Comput. Mater. 2019, 5, 84.
- Venkatraman, V. The utility of composition-based machine learning models for band gap prediction. Comput. Mater. Sci. 2021, 197, 110637.
- Xu, P.; Lu, T.; Ju, L.; Tian, L.; Li, M.; Lu, W. Machine Learning Aided Design of Polymer with Targeted Band Gap Based on DFT Computation. J. Phys. Chem. B 2021, 125, 601–611.
- Espinosa, R.; Ponce, H.; Ortiz-Medina, J. A 3D orthogonal vision-based band-gap prediction using deep learning: A proof of concept. Comput. Mater. Sci. 2022, 202, 110967.
- Wang, T.; Zhang, K.; Thé, J.; Yu, H. Accurate prediction of band gap of materials using stacking machine learning model. Comput. Mater. Sci. 2022, 201, 110899.
- Na, G.S.; Jang, S.; Lee, Y.-L.; Chang, H. Tuplewise material representation based machine learning for accurate band gap prediction. J. Phys. Chem. A 2020, 124, 10616–10623.
- Shen, Z.H.; Liu, H.X.; Shen, Y.; Hu, J.M.; Chen, L.Q.; Nan, C.W. Machine learning in energy storage materials. Interdiscip. Mater. 2022, 1, 175–195.
- Feng, Y.; Tang, W.; Zhang, Y.; Zhang, T.; Shang, Y.; Chi, Q.; Chen, Q.; Lei, Q. Machine learning and microstructure design of polymer nanocomposites for energy storage application. High Volt. 2022, 7, 242–250.
- Yue, D.; Feng, Y.; Liu, X.X.; Yin, J.H.; Zhang, W.C.; Guo, H.; Su, B.; Lei, Q.Q. Prediction of Energy Storage Performance in Polymer Composites Using High-Throughput Stochastic Breakdown Simulation and Machine Learning. Adv. Sci. 2022, 9, 2105773.
- Ojih, J.; Onyekpe, U.; Rodriguez, A.; Hu, J.; Peng, C.; Hu, M. Machine Learning Accelerated Discovery of Promising Thermal Energy Storage Materials with High Heat Capacity. ACS Appl. Mater. Interfaces 2022, 14, 43277–43289.
- Malekloo, A.; Ozer, E.; AlHamaydeh, M.; Girolami, M. Machine learning and structural health monitoring overview with emerging technology and high-dimensional data source highlights. Struct. Health Monit. 2022, 21, 1906–1955.
- Cao, Y.; Miraba, S.; Rafiei, S.; Ghabussi, A.; Bokaei, F.; Baharom, S.; Haramipour, P.; Assilzadeh, H. Economic application of structural health monitoring and internet of things in efficiency of building information modeling. Smart Struct. Syst. 2020, 26, 559–573.
- Dang, H.V.; Tatipamula, M.; Nguyen, H.X. Cloud-based digital twinning for structural health monitoring using deep learning. IEEE Trans. Ind. Inform. 2021, 18, 3820–3830.
- Dong, W.; Huang, Y.; Lehane, B.; Ma, G. XGBoost algorithm-based prediction of concrete electrical resistivity for structural health monitoring. Autom. Constr. 2020, 114, 103155.
- Fu, B.; Chen, S.-Z.; Liu, X.-R.; Feng, D.-C. A probabilistic bond strength model for corroded reinforced concrete based on weighted averaging of non-fine-tuned machine learning models. Constr. Build. Mater. 2022, 318, 125767.
- Gao, J.; Koopialipoor, M.; Armaghani, D.J.; Ghabussi, A.; Baharom, S.; Morasaei, A.; Shariati, A.; Khorami, M.; Zhou, J. Evaluating the bond strength of FRP in concrete samples using machine learning methods. Smart Struct. Syst. Int. J. 2020, 26, 403–418.
- Li, Z.; Qi, J.; Hu, Y.; Wang, J. Estimation of bond strength between UHPC and reinforcing bars using machine learning approaches. Eng. Struct. 2022, 262, 114311.
- Su, M.; Zhong, Q.; Peng, H.; Li, S. Selected machine learning approaches for predicting the interfacial bond strength between FRPs and concrete. Constr. Build. Mater. 2021, 270, 121456.
- Khan, B.M.; Cohen, Y. Predictive Nanotoxicology: Nanoinformatics Approach to Toxicity Analysis of Nanomaterials. In Machine Learning in Chemical Safety and Health: Fundamentals with Applications; John Wiley & Sons: Hoboken, NJ, USA, 2022; pp. 199–250.
- Huang, Y.; Li, X.; Cao, J.; Wei, X.; Li, Y.; Wang, Z.; Cai, X.; Li, R.; Chen, J. Use of dissociation degree in lysosomes to predict metal oxide nanoparticle toxicity in immune cells: Machine learning boosts nano-safety assessment. Environ. Int. 2022, 164, 107258.
- Gousiadou, C.; Marchese Robinson, R.; Kotzabasaki, M.; Doganis, P.; Wilkins, T.; Jia, X.; Sarimveis, H.; Harper, S. Machine learning predictions of concentration-specific aggregate hazard scores of inorganic nanomaterials in embryonic zebrafish. Nanotoxicology 2021, 15, 446–476.
- Liu, L.; Zhang, Z.; Cao, L.; Xiong, Z.; Tang, Y.; Pan, Y. Cytotoxicity of phytosynthesized silver nanoparticles: A meta-analysis by machine learning algorithms. Sustain. Chem. Pharm. 2021, 21, 100425.
- Sajid, M.; Ihsanullah, I.; Khan, M.T.; Baig, N. Nanomaterials-based adsorbents for remediation of microplastics and nanoplastics in aqueous media: A review. Sep. Purif. Technol. 2022, 305, 122453.
- Moosavi, S.; Manta, O.; El-Badry, Y.A.; Hussein, E.E.; El-Bahy, Z.M.; Mohd Fawzi, N.f.B.; Urbonavičius, J.; Moosavi, S.M.H. A study on machine learning methods’ application for dye adsorption prediction onto agricultural waste activated carbon. Nanomaterials 2021, 11, 2734.
- Guo, W.; Liu, J.; Dong, F.; Chen, R.; Das, J.; Ge, W.; Xu, X.; Hong, H. Deep learning models for predicting gas adsorption capacity of nanomaterials. Nanomaterials 2022, 12, 3376.
- Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Duvenaud, D.; Maclaurin, D.; Blood-Forsythe, M.A.; Chae, H.S.; Einzinger, M.; Ha, D.-G.; Wu, T. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 2016, 15, 1120–1127.
- Xue, D.; Yuan, R.; Zhou, Y.; Xue, D.; Lookman, T.; Zhang, G.; Ding, X.; Sun, J. Design of high temperature Ti-Pd-Cr shape memory alloys with small thermal hysteresis. Sci. Rep. 2016, 6, 28244.
- Li, W.; Yang, T.; Liu, C.; Huang, Y.; Chen, C.; Pan, H.; Xie, G.; Tai, H.; Jiang, Y.; Wu, Y. Optimizing piezoelectric nanocomposites by high-throughput phase-field simulation and machine learning. Adv. Sci. 2022, 9, 2105550.
- Zhang, L.; He, M.; Shao, S. Machine learning for halide perovskite materials. Nano Energy 2020, 78, 105380.
- Li, L.; Tao, Q.; Xu, P.; Yang, X.; Lu, W.; Li, M. Studies on the regularity of perovskite formation via machine learning. Comput. Mater. Sci. 2021, 199, 110712.
- Liu, H.; Cheng, J.; Dong, H.; Feng, J.; Pang, B.; Tian, Z.; Ma, S.; Xia, F.; Zhang, C.; Dong, L. Screening stable and metastable ABO3 perovskites using machine learning and the materials project. Comput. Mater. Sci. 2020, 177, 109614.
- Omprakash, P.; Manikandan, B.; Sandeep, A.; Shrivastava, R.; Viswesh, P.; Panemangalore, D.B. Graph representational learning for bandgap prediction in varied perovskite crystals. Comput. Mater. Sci. 2021, 196, 110530.
- Wang, Z.; Cai, J.; Wang, Q.; Wu, S.; Li, J. Unsupervised discovery of thin-film photovoltaic materials from unlabeled data. NPJ Comput. Mater. 2021, 7, 128.
- Huang, K.; Zhan, X.-L.; Chen, F.-Q.; Lü, D.-W. Catalyst design for methane oxidative coupling by using artificial neural network and hybrid genetic algorithm. Chem. Eng. Sci. 2003, 58, 81–87.
- Zhang, S.; Lu, S.; Zhang, P.; Tian, J.; Shi, L.; Ling, C.; Zhou, Q.; Wang, J. Accelerated Discovery of Single-Atom Catalysts for Nitrogen Fixation via Machine Learning. Energy Environ. Mater. 2023, 6, e12304.
- Wei, S.; Baek, S.; Yue, H.; Liu, M.; Yun, S.J.; Park, S.; Lee, Y.H.; Zhao, J.; Li, H.; Reyes, K. Machine-learning assisted exploration: Toward the next-generation catalyst for hydrogen evolution reaction. J. Electrochem. Soc. 2021, 168, 126523.
- Hueffel, J.A.; Sperger, T.; Funes-Ardoiz, I.; Ward, J.S.; Rissanen, K.; Schoenebeck, F. Accelerated dinuclear palladium catalyst identification through unsupervised machine learning. Science 2021, 374, 1134–1140.
- Zhang, J.; Zhu, Z.; Xiang, X.-D.; Zhang, K.; Huang, S.; Zhong, C.; Qiu, H.-J.; Hu, K.; Lin, X. Machine learning prediction of superconducting critical temperature through the structural descriptor. J. Phys. Chem. C 2022, 126, 8922–8927.
- Le, T.D.; Noumeir, R.; Quach, H.L.; Kim, J.H.; Kim, J.H.; Kim, H.M. Critical temperature prediction for a superconductor: A variational bayesian neural network approach. IEEE Trans. Appl. Supercond. 2020, 30, 8600105.
- Zhang, J.; Zhang, K.; Xu, S.; Li, Y.; Zhong, C.; Zhao, M.; Qiu, H.-J.; Qin, M.; Xiang, X.-D.; Hu, K. An integrated machine learning model for accurate and robust prediction of superconducting critical temperature. J. Energy Chem. 2023, 78, 232–239.
- Roter, B.; Dordevic, S. Predicting new superconductors and their critical temperatures using machine learning. Phys. C Supercond. Its Appl. 2020, 575, 1353689.
- Pereti, C.; Bernot, K.; Guizouarn, T.; Laufek, F.; Vymazalová, A.; Bindi, L.; Sessoli, R.; Fanelli, D. From individual elements to macroscopic materials: In search of new superconductors via machine learning. NPJ Comput. Mater. 2023, 9, 71.
- Xie, Y.; Zhang, C.; Hu, X.; Zhang, C.; Kelley, S.P.; Atwood, J.L.; Lin, J. Machine learning assisted synthesis of metal–organic nanocapsules. J. Am. Chem. Soc. 2019, 142, 1475–1481.
- Pellegrino, F.; Isopescu, R.; Pellutiè, L.; Sordello, F.; Rossi, A.M.; Ortel, E.; Martra, G.; Hodoroaba, V.-D.; Maurino, V. Machine learning approach for elucidating and predicting the role of synthesis parameters on the shape and size of TiO2 nanoparticles. Sci. Rep. 2020, 10, 18910.
- Tao, H.; Wu, T.; Aldeghi, M.; Wu, T.C.; Aspuru-Guzik, A.; Kumacheva, E. Nanoparticle synthesis assisted by machine learning. Nat. Rev. Mater. 2021, 6, 701–716.
- Braham, E.J.; Cho, J.; Forlano, K.M.; Watson, D.F.; Arròyave, R.; Banerjee, S. Machine learning-directed navigation of synthetic design space: A statistical learning approach to controlling the synthesis of perovskite halide nanoplatelets in the quantum-confined regime. Chem. Mater. 2019, 31, 3281–3292.
- Epps, R.W.; Bowen, M.S.; Volk, A.A.; Abdel-Latif, K.; Han, S.; Reyes, K.G.; Amassian, A.; Abolhasani, M. Artificial chemist: An autonomous quantum dot synthesis bot. Adv. Mater. 2020, 32, 2001626.
- Wang, J.; Wang, Y.; Chen, Y. Inverse design of materials by machine learning. Materials 2022, 15, 1811.
- Li, S.; Barnard, A.S. Inverse Design of Nanoparticles Using Multi-Target Machine Learning. Adv. Theory Simul. 2022, 5, 2100414.
- Wang, R.; Liu, C.; Wei, Y.; Wu, P.; Su, Y.; Zhang, Z. Inverse design of metal nanoparticles based on deep learning. Results Opt. 2021, 5, 100134.
- He, J.; He, C.; Zheng, C.; Wang, Q.; Ye, J. Plasmonic nanoparticle simulations and inverse design using machine learning. Nanoscale 2019, 11, 17444–17459.
This entry is offline, you can click here to edit this entry!