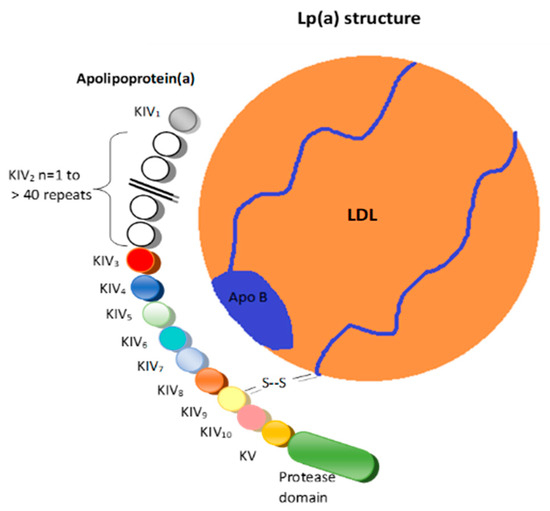
Apolipoprotein(a) (apo(a)) is the protein component that defines lipoprotein(a) (Lp(a)) particles and is encoded by the LPA gene. The apo(a) is extremely heterogeneous in size due to the copy number variation (CNV) in the kringle IV type-2 (KIV-2) domains. Lp(a) concentrations in the blood vary by more than a thousand-fold between individuals, ranging from less than 0.1 to more than 300 mg/dL, depending on the size of apo(a) that is encoded by the LPA gene. The KIV-2 copy number ranges from 1 to >40, and the CNV of KIV-2 shows a >95% heterozygosity in most populations. Screening patients for elevated Lp(a) is strongly encouraged as an effective tool to identify individuals requiring more aggressive lipid-lowering therapy to reduce the CVD risk. Lp(a) levels above 50 mg/dL are correlated with an increased risk for the development of CVD.
- lipoprotein(a)
- apolipoprotein(a)
- LPA gene
- KIV-2
- CNV
- genotyping
- phenotyping
1. Introduction
2. Genotyping Techniques
2.1. Pulsed-Field Gel Electrophoresis (PFGE)/Southern Blot
2.2. Quantitative Polymerase Chain Reaction (qPCR)
2.3. Fiber Fluorescence In Situ Hybridization (Fiber-FISH)
3. Phenotyping Techniques
Western Blot Using Antibodies for apo(a) (Immunoblotting)
This entry is adapted from the peer-reviewed paper 10.3390/ijms241813886
References
- Koschinsky, M.L.; Kronenberg, F. The long journey of lipoprotein(a) from cardiovascular curiosity to therapeutic target. Atherosclerosis 2022, 349, 1–6.
- Arsenault, B.J.; Kamstrup, P.R. Lipoprotein(a) and cardiovascular and valvular diseases: A genetic epidemiological perspective. Atherosclerosis 2022, 349, 7–16.
- Wang, H.; Wu, P.; Jiang, D.; Zhang, H.; Zhang, J.; Zong, Y.; Han, Y. Relationship between serum homocysteine, fibrinogen, lipoprotein-a level, and peripheral arterial disease: A dose-response meta-analysis. Eur. J. Med. Res. 2022, 27, 261.
- Singh, S.; Baars, D.P.; Desai, R.; Singh, D.; Pinto-Sietsma, S.J. Association between Lipoprotein(a) and risk of atrial fibrillation: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. Curr. Probl. Cardiol. 2023, 49, 102024.
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91.
- Marcovina, S.M.; Morrisett, J.D. Structure and metabolism of lipoprotein(a). Curr. Opin. Lipidol. 1995, 6, 136–145.
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35.
- Fogacci, F.; Cicero, A.F.G.; D’Addato, S.; Giovannini, M.; Borghi, C.; Brisighella Heart Study Group. Effect of spontaneous changes in dietary components and lipoprotein(a) levels: Data from the Brisighella Heart Study. Atherosclerosis 2017, 262, 202–204.
- Matveyenko, A.; Matienzo, N.; Ginsberg, H.; Nandakumar, R.; Seid, H.; Ramakrishnan, R.; Holleran, S.; Thomas, T.; Reyes-Soffer, G. Relationship of apolipoprotein(a) isoform size with clearance and production of lipoprotein(a) in a diverse cohort. J. Lipid. Res. 2023, 64, 100336.
- Cicero, A.F.G.; Fogacci, F.; Derosa, G.; D’Angelo, A.; Ventura, F.; Rizzoli, E.; D’Addato, S.; Borghi, C.; On Behalf of The Brisighella Heart Study Group. Lipoprotein(a) Serum Levels Predict Pulse Wave Velocity in Subjects in Primary Prevention for Cardiovascular Disease with Large Apo(a) Isoforms: Data from the Brisighella Heart Study. Biomedicines 2022, 10, 656.
- Grüneis, R.; Lamina, C.; Di Maio, S.; Schönherr, S.; Zoescher, P.; Forer, L.; Streiter, G.; Peters, A.; Gieger, C.; Köttgen, A.; et al. The effect of LPA Thr3888Pro on lipoprotein(a) and coronary artery disease is modified by the LPA KIV-2 variant 4925G>A. Atherosclerosis 2022, 349, 151–159.
- Sandholzer, C.; Saha, N.; Kark, J.D.; Rees, A.; Jaross, W.; Dieplinger, H.; Hoppichler, F.; Boerwinkle, E.; Utermann, G. Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler. Thromb. 1992, 12, 1214–1226.
- Tsarouhas, K.; Hoursalas, A.; Vardavas, A.I.; Tsitsimpikou, C. Lipoprotein a: An emerging risk identifier and evolving clinical target. Public Health Toxicol. 2022, 2, 2.
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166.
- Zysow, B.R.; Lindahl, G.E.; Wade, D.P.; Knight, B.L.; Lawn, R.M. C/T polymorphism in the 5’ untranslated region of the apolipoprotein(a) gene introduces an upstream ATG and reduces in vitro translation. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 58–64.
- Gu, J.X.; Huang, J.; Li, S.S.; Zhou, L.H.; Yang, M.; Li, Y.; Zhang, A.M.; Yin, Y.; Zhang, N.; Jia, M.; et al. Elevated lipoprotein(a) and genetic polymorphisms in the LPA gene may predict cardiovascular events. Sci. Rep. 2022, 12, 3588.
- Kraft, H.G.; Köchl, S.; Menzel, H.J.; Sandholzer, C.; Utermann, G. The apolipoprotein (a) gene: A transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet. 1992, 90, 220–230.
- Anglés-Cano, E.; Loyau, S.; Cardoso-Saldaña, G.; Couderc, R.; Gillery, P. A novel kringle-4 number-based recombinant apo standard for human apo phenotyping. J. Lipid. Res. 1999, 40, 354–359.
- Lackner, C.; Boerwinkle, E.; Leffert, C.C.; Rahmig, T.; Hobbs, H.H. Molecular basis of apolipoprotein (a) isoform size hetero-geneity as revealed by pulsed-field gel electrophoresis. J. Clin. Investig. 1991, 87, 2153–2161.
- Chiou, C.S.; Wei, H.L.; Yang, L.C. Comparison of pulsed-field gel electrophoresis and coagulase gene restriction profile analysis techniques in the molecular typing of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 2186–2190.
- Li, Y.; Wang, Y.; Gong, F.; Yu, X.; Zhang, T. A novel deletion mutation in the LPA gene in a middle-aged woman with ischaemic stroke. BMC Med. Genom. 2021, 14, 132.
- Lanktree, M.B.; Rajakumar, C.; Brunt, J.H.; Koschinsky, M.L.; Connelly, P.W.; Hegele, R.A. Determination of lipoprotein(a) krin-gle repeat number from genomic DNA: Copy number variation genotyping using qPCR. J. Lipid. Res. 2009, 50, 768–772.
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid. Res. 2016, 57, 1339–1359.
- Kraft, H.G.; Lingenhel, A.; Köchl, S.; Hoppichler, F.; Kronenberg, F.; Abe, A.; Mühlberger, V.; Schönitzer, D.; Utermann, G. Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 713–719.
- Wild, S.H.; Fortmann, S.P.; Marcovina, S.M. A prospective case-control study of lipoprotein(a) levels and apo(a) size and risk of coronary heart disease in Stanford Five-City Project participants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 239–245.
- Erdel, M.; Hubalek, M.; Lingenhel, A.; Kofler, K.; Duba, H.-C.; Utermann, G. Counting the repetitive kringle-IV repeats in the gene encoding human apolipoprotein(a) by fibre-FISH. Nat. Genet. 1999, 21, 357–358.
- Enkhmaa, B.; Anuurad, E.; Zhang, W.; Tran, T.; Berglund, L. Lipoprotein(a): Genotype-phenotype relationship and impact on atherogenic risk. Metab. Syndr. Relat. Disord. 2011, 9, 411–418.
- Pradhan, S.; Apaydin, S.; Bucevičius, J.; Gerasimaitė, R.; Kostiuk, G.; Lukinavičius, G. Sequence-specific DNA labelling for fluorescence microscopy. Biosens. Bioelectron 2023, 230, 115256.
- Zekavat, S.M.; Ruotsalainen, S.; Handsaker, R.E.; Alver, M.; Bloom, J.; Poterba, T.; Seed, C.; Ernst, J.; Chaffin, M.; Engreitz, J.; et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat. Commun. 2018, 9, 2606.
- Mazur, P.; Dumnicka, P.; Tisończyk, J.; Ząbek-Adamska, A.; Drożdż, R. SDS Electrophoresis on Gradient Polyacrylamide Gels as a Semiquantitative Tool for the Evaluation of Proteinuria. Diagnostics 2023, 13, 1513.
- Koschinsky, M.L.; Beisiegel, U.; Henne-Bruns, D.; Eaton, D.L.; Lawn, R.M. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry 1990, 29, 640–644.
- Kamboh, M.I.; Ferrell, R.E.; Kottke, B.A. Expressed hypervariable polymorphism of apolipoprotein (a). Am. J. Hum. Genet. 1991, 49, 1063–1074.
- Geroldi, D.; Bellotti, V.; Buscaglia, P.; Bonetti, G.; Gazzaruso, C.; Caprioli, A.; Fratino, P. Characterization of apo(a) polymorphism by a modified immunoblotting technique in an Italian population sample. Clin. Chim. Acta 1993, 221, 159–169.