Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pharmacology & Pharmacy
Post-traumatic stress disorder (PTSD) is a psychopathological condition with a heterogeneous clinical picture that is complex and challenging to treat. Its multifaceted pathophysiology still remains an unresolved question and certainly contributes to this issue. The pharmacological treatment of PTSD is mainly empirical and centered on the serotonergic system. Since the therapeutic response to prescribed drugs targeting single symptoms is generally inconsistent, there is an urgent need for novel pathogenetic hypotheses, including different mediators and pathways.
- PTSD
- pharmacological treatment
- hypothalamic-pituitary-adrenal axis
- opioids
- glutamate
- cannabinoids
- oxytocin
- microRNA
1. Introduction
Post-traumatic stress disorder (PTSD) is a psychiatric condition resulting from exposure to severe stressful events threatening one’s or others’ lives or physical integrity, experienced with intense distress, fear, and horror, and characterized by a multiplicity of symptoms [1]. Initially described following the first 19th-century railway disasters, PTSD has been mainly defined and studied in the USA after the first and second world wars, but especially among the veterans of the Vietnam War [2][3][4]. The epidemiological data on PTSD are variable, as they often reflect different and/or specific population groups; in any case, its prevalence is estimated to range between 5% and 10% [5], while being higher in the female sex [6][7][8] and in the military corps, where it may increase to 16.8% [9]. Post-traumatic stress disorder shows a wide range of potential symptoms, including affective and cognitive disturbances such as negative beliefs of the self or of the world, persistently depressed mood, anhedonia, feelings of estrangement or detachment from others, and an inability to experience positive emotions. Again, patients with PTSD may also suffer from hyperarousal, persistent psychological distress in response to triggers, an avoidance of places/situations reminiscent of the traumatic event, flashbacks and dissociative experiences, irritability, anger outbursts, and reckless or self-destructive behavior [10]. Furthermore, PTSD may be comorbid with mood, anxiety, and substance use disorders [1], and it may also be associated with high suicide rates [11]. Interestingly, between 3.6 and 25.6% of people may suffer from PTSD symptoms that, although not fully satisfying the criteria of the main diagnostic systems, generally go unnoticed in clinical care settings but would require medical attention since they may cause discomfort and lead to decreased family, social, and work maladjustment, and even depression with suicidal thoughts [12]. Although the pathophysiology of PTSD is largely unknown, the current biological hypotheses underline the key role of the brain circuitry regulating fear processes involving the amygdala, insula, hippocampus, and medial prefrontal cortex and related neurotransmitters, as well as the stress system that is the hypothalamic-pituitary adrenal axis (HPA) [13]. Not surprisingly, the pharmacological approach to PTSD is largely empirical and centered on the serotonin (5-HT) system, which constitutes the main target of current treatments. Nevertheless, it is evident that its multifaceted clinical picture would suggest that other systems and pathways are undoubtedly involved in its pathophysiology. In addition, the evidence that a traumatic event may not systematically lead to the development of PTSD or a trauma-related disorder strongly suggests that this condition results from the individual’s vulnerability, possibly on a genetic and/or epigenetic basis intertwined with environmental factors. Needless to say, there is an urgent need for tailored and targeted drugs to manage PTSD. If more recent data strengthen the involvement of the HPA axis and stress processes, preliminary evidence suggests that the opioid peptides, glutamate, cannabinoids, oxytocin, neuropeptide Y, and microRNA would be involved in PTSD pathophysiology [14][15][16][17][18].
2. The Hypothalamic-Pituitary-Adrenal Axis
It is well known that the HPA plays an important role in the response to stress through the corticotropin-releasing factor (CRF) acting on CRF1 receptors that are present throughout the whole brain but mainly in the hypothalamus and amygdala [19] (Figure 1). Clinical data indicate that dysfunction of the stress response system, excessive CRF activity, and possible excessive stimulation of CRF1 receptors are present in a wide range of stress-related disorders such as depression, anxiety, irritable bowel syndrome [20], and, not surprisingly, PTSD. The CRF1 receptor alteration might be particularly relevant in the most severe forms of these conditions, e.g., melancholic or psychotic depression or chronic PTSD, and/or when they are accompanied by a history of early life trauma [21][22][23]. Preclinical studies demonstrate that CRF1 receptor antagonists are effective in animal models in which CRF pathways and CRF1 receptors are hyperactivated, whereas they tend to be quiescent in states of low basal CRF activity, thus indicating potentially reduced side effects in humans [24]. Symptom heterogeneity in animal models of stress and in human stress disorders might result from dysfunctions in different CRF1 receptor populations and/or functional states. Blood cortisol concentrations in PTSD patients reveal larger 24-h fluctuations than normal or depressed controls. At variance with normal states, this and the noradrenergic systems seem to stimulate each other, supporting the hypothesis of deranged negative feedback between these two systems in PTSD [17].
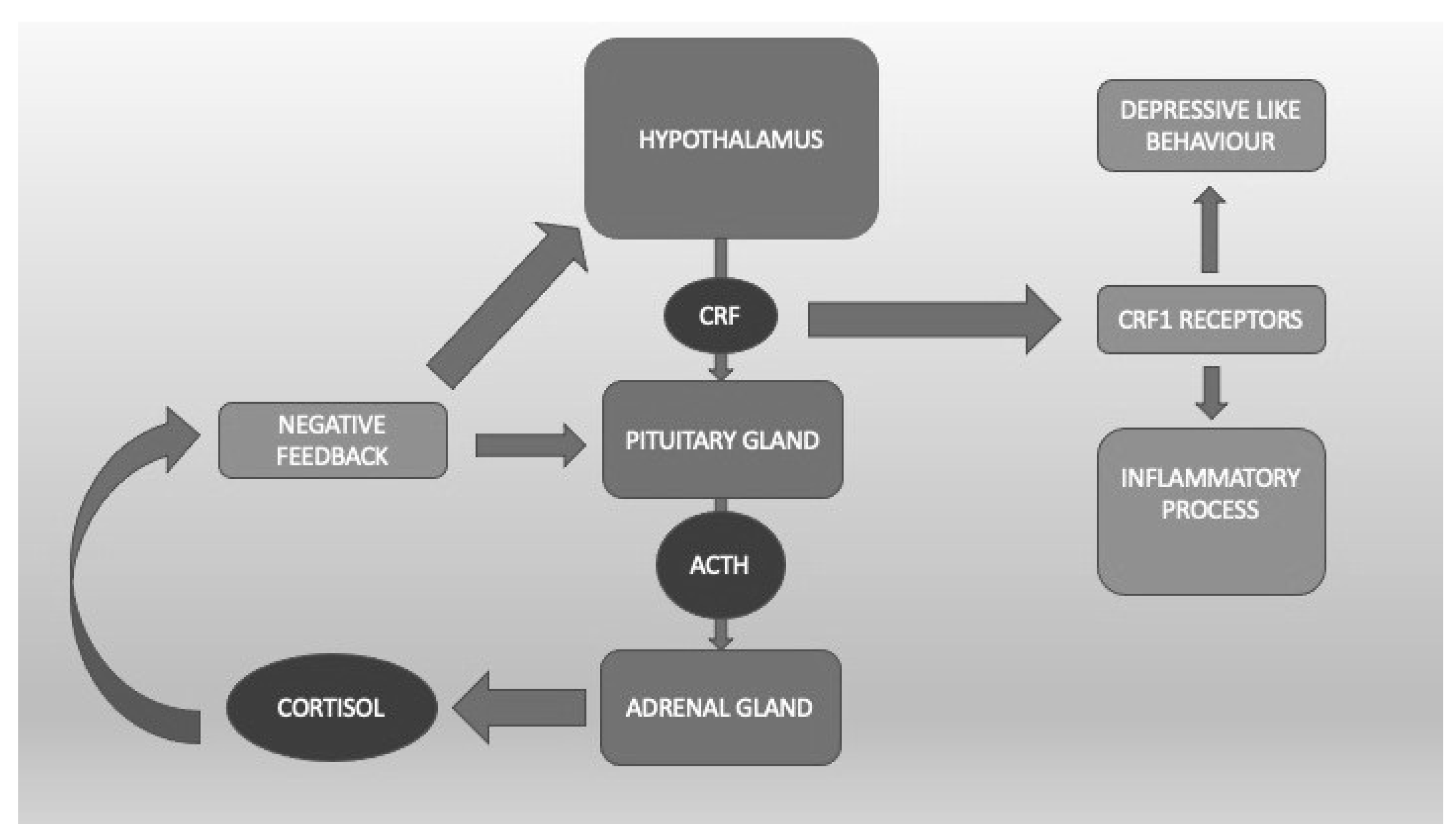
Figure 1. The hypothalamic-pituitary-adrenal axis. Corticotropin Releasing Factor (CRF) binds CRF1 receptors, which are linked to the inflammatory process and depressive-like behavior.
To summarize, these data suggesting the eventual clinical utility of the potential of CRF1 receptor modulation or HPA dampening by neurosteroids in PTSD treatment are interesting and cannot be ignored, in spite of being limited (Figure 1).
3. Opioid Peptides
The opioid peptide family includes beta-endorphins, enkephalins, and dynorphins. The first two are present in the hypothalamus, anterior and intermediate pituitary, diencephalon, pons, and locus coeruleus; the second ones are in the hypothalamus, basal ganglia, hippocampus, medullary raphe, nucleus accumbens, amygdala, and pons. Dynorphin A and B are located in the hypothalamus, dentate gyrus, periaqueductal gray, cortex, hippocampus, olfactory bulb, globus pallidus, substantia nigra, and putamen. Opioids act on three classical receptors coupled to G proteins, μ, δ, κ opioid receptors (MOR, DOR, and KOR, respectively), and on the 1/nociceptin receptor, similar to the non-classical opioid receptor [25]. It is known that the activation of each of these receptors is associated with several outcomes [26]. The MOR and DOR are linked to central anesthesia and respiratory depression, while the KOR is linked to peripheral anesthesia but not respiratory depression. The MOR is related to euphoria, while the KOR is related to dysphoria. The MOR and KOR may provoke sedation, and the only MOR has tolerance and abstinence phenomena [27].
Disturbances in the regulation of endogenous opioids might be involved in some symptoms of PTSD, such as freezing, stress-induced analgesia, and dissociation [28]. Therefore, potentially, opioid peptides might play a role in PTSD treatment. Some clinical evidence suggests that administration of morphine immediately after the traumatic event may reduce the likelihood of developing PTSD, although the precise mechanism of this effect is unknown [29][30]. It has been hypothesized that it may be due to an indirect decrease in noradrenergic activity or to a direct action on the interneurons of the amygdala that are critical for fear extinction processes [31]. Veterans suffering from PTSD are more likely to receive opioid prescriptions and higher or more opioid doses for the treatment of chronic pain [32]. If, from one point of view, the prescription practice contributes to a high consumption of opioids among veterans [33][34], it is well known that acute opioid administration following trauma is protective and, as already mentioned, may reduce the likelihood of developing PTSD [29][30]. Patients receiving lower doses of morphine in the first 48 h after a traumatic injury more frequently developed PTSD than patients receiving higher doses [30]. Similar results have been found in children who received morphine for burns [35]. This may be related to the effect of pain on the outcomes of PTSD. The level of pain felt at the time of a traumatic injury is predictive of the development of PTSD [36]. Another contributing factor may be the effect of morphine on long-term memory [37]. Several opioids can vary greatly in their effects, such as their ability to suppress pain, as well as in their differential risks of abuse, respiratory depression, and worsening/precipitation comorbidities with other psychological disorders [26]. Buprenorphine, a partial agonist at MOR and an antagonist at KOR, seems to show greater beneficial effects than complete agonists in the treatment of PTSD patients [38].
It is evident that the opioid system is widely involved in major PTSD symptoms and that opioid compounds may be effective. However, the abuse potential of these drugs, as reported among military groups and veterans, requires caution in their large-scale use, at least until the development of novel and less dangerous compounds.
4. Glutamate
Glutamate is an excitatory neurotransmitter present in pyramidal cells and in most brain synapses, where it counterbalances the activity of the major inhibitory neurotransmitter (GABA) [39], which is one of its products through the glutamate decarboxylase. Glutamate binds to ionotropic glutamate receptors (iGluRs) that are cation-permeable ligand-gated ion channels and metabotropic glutamate receptors (mGluRs) that are G protein-coupled receptors. The activation of mGluRs and iGluRs results in distinct cellular reactions. The iGluRs are split into several functional classes, including GluD receptors (also known as delta receptors), kainate receptors, N-methyl-d-aspartate (NMDA) receptors, and -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [40] (Figure 2). Glutamate plays an important role in synaptic plasticity and, therefore, in cognitive functions such as learning and memory [41][42].
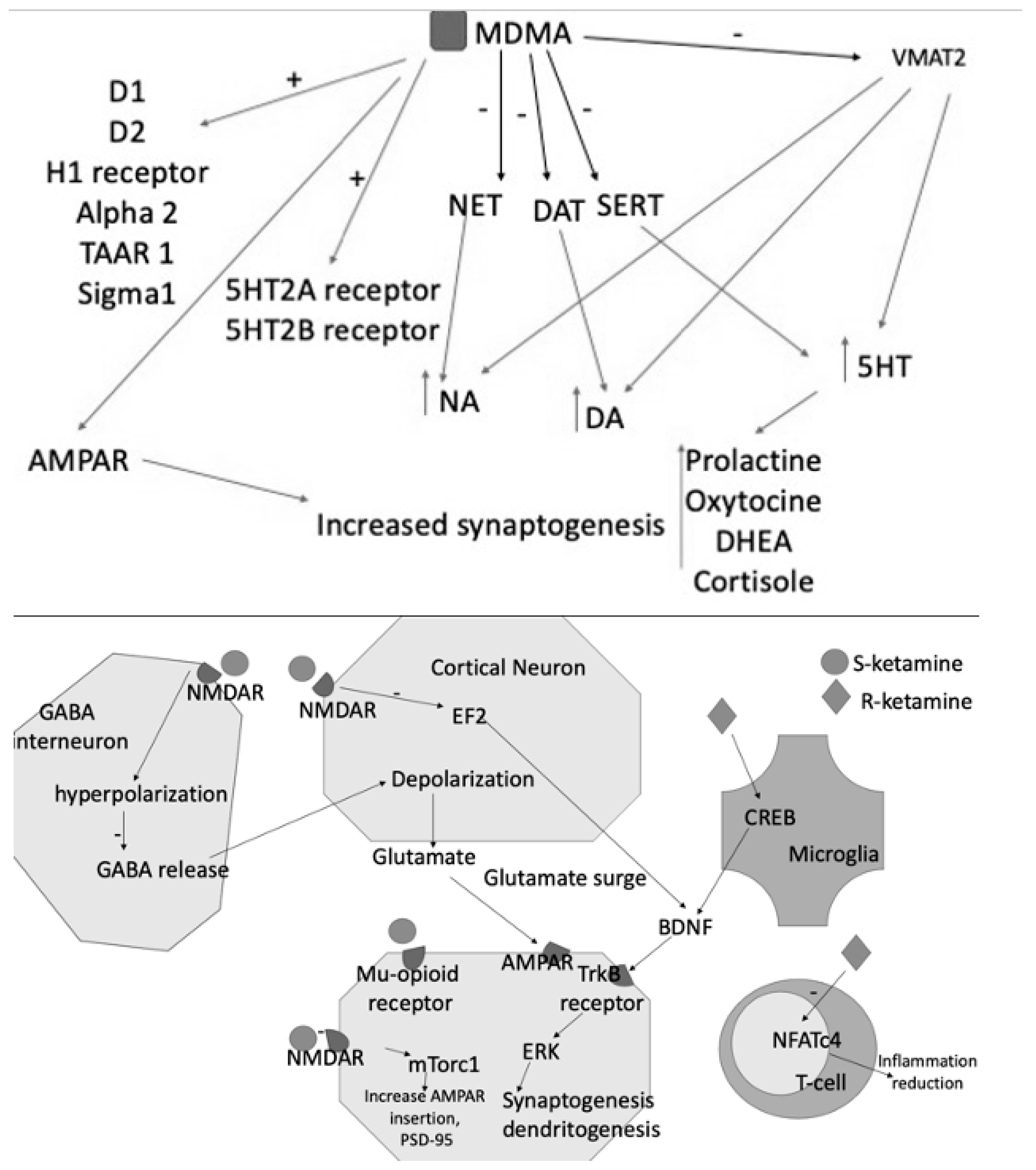
Figure 2. MDMA acts as an inhibitor of NET, DAT, SERT and of VMAT2, resulting in increased levels of the three monoamines DA, NE, and 5HT. The latter consequently increases the blood levels of prolactin, DHEA, oxytocin, and cortisol. MDMA also binds D1-D2 receptors and acts as an agonist of H1, 5HT2A, and B receptors. Its effects are also explained by the interaction with TAAR1, alpha2, and Sigma1. S- and R-ketamines increase glutamate release into the synaptic cleft, activating AMPAR and contributing to further synaptogenesis and dendritogenesis. S-ketamine binds NMDARs expressed in GABAergic interneurons, leading to a depolarization of cortical excitatory neurons. This depolarization causes glutamate and BDNF release, which bind to TrkB receptors. TrkB activates the mTORC1 signaling pathway, leading to the upregulation of synaptogenesis and dendritogenesis. S-ketamine binds to extrasynaptic NMDARs, disinhibiting mTORC1 signaling by deactivating eEFK2. Binding to mu-opioid receptors may facilitate antidepressant effects. R-ketamine affects microglial signaling and, by increasing BDNF release through TrkB, activates the ERK signaling pathway, resulting in synaptogenesis and dendritogenesis.
Similarly, psilocybin, a naturally occurring psychedelic compound, has been recently rediscovered for its potential therapeutic effects, particularly as a potential alternative antidepressant in MDD [43]. A preclinical study reported the anti-inflammatory effects of psilocybin, alone and in combination with eugenol, in the brain of mice with systemically induced inflammation, suggesting a potential mechanism through which psilocybin could exert its therapeutic effects in disorders associated with elevated inflammatory processes, such as PTSD [44]. However, to date, there is only one open-label pilot study, which is currently exploring the safety and efficacy of psilocybin-assisted therapy among U.S. Military veterans with severe, treatment-resistant PTSD. In particular, this research aims at investigating the combination of two psilocybin administration sessions with psychotherapy and addressing the limitations of current PTSD treatments, especially within the veteran population [45].
Taken together, the preliminary findings that glutamate modulation might constitute a possible therapeutic strategy for PTSD appear quite intriguing, even considering that esketamine is already marketed for treatment-resistant depression. Other compounds seem to show a certain degree of effectiveness that, however, needs to be substantiated in controlled clinical trials (Figure 2).
5. Cannabinoids
The cannabinoid (CBD) system, including endogenous cannabinoids, endocannabinoid production and degradation enzymes, and cannabinoid receptors, has been recently suggested to be involved in the pathophysiology of PTSD [14]. Anandamide (arachidonoyl ethanolamide) and 2-arachidonoyl glycerol (2-AG) are the first identified and well-defined endocannabinoids deriving from lipid membranes. They exert their activity through two G protein-coupled receptors called CB1 and CB2. The CB1 receptors are more abundant in the CNS than the CB2 receptors. The cortex, hippocampus, basal ganglia, and cerebellum are particularly rich in CB1 receptors, mainly on axon terminals and pre-terminal axon segments. Cortical and hippocampal CB1 receptors that are typically expressed at lower levels in glutamatergic neurons are notably concentrated in cholecystokinin (CCK)-positive interneurons. The CB1 receptors are also found in basket cells, parallel fibers, and climbing fibers in the cerebellum. Despite being identified on many neurons, CB1 receptors are also found in glial cells, and vascular tissues and microglia include this receptor. However, it is thought that some neurons express CB2, particularly in pathological circumstances such as nerve damage. A particularly intriguing property is the ability of CB2 receptor expression to rise up to 100 times following tissue injury or during inflammation. It is still unclear whether the observed increases in CB2 in the CNS are the consequence of immune cells from the periphery moving into the CNS or increased CB2 expression on CNS cells.
When required, endocannabinoids are quickly released into the extracellular space through one or two enzymatic processes (usually induced by activation of certain G protein-coupled receptors or by depolarization). The intrinsic activity of endogenous cannabinoids is different: 2-AG behaves as a high agonist at both CB1 and CB2 receptors, while anandamide shows a low and very low effect at, respectively, CB1 and CB2 receptors. Therefore, in systems with low receptor expression or when receptors connect to signaling pathways in a weak way, anandamide can counterbalance the effects of more powerful agonists. Additional endogenous substances (such as virodhamine) may increase the activity of other endocannabinoids, but the biology of these substances is less understood than that of anandamide and 2-AG. Although the effects of endocannabinoids are mostly mediated through CB1 and CB2 receptors, additional receptors, such as transient receptor potential (TRP) channels and peroxisome proliferator-activated receptor (PPAR), are supposed to mediate some endocannabinoid functions. The activation of CB1 or CB2 receptors affects cellular physiology in a variety of ways, altering gene transcription, cell motility, and synaptic function. In some circumstances, anandamide opens transient potential receptor (TRP) channels, especially TRPV1. Anandamide also activates PPAR alpha and gamma, which have a significant effect on gene transcription. It is important to keep in mind that boosting anandamide levels by blocking fatty acid amide hydrolase (FAAH) breakdown also increases concentrations of other N-acylamides that may affect PPAR [46].
6. Oxytocin
Oxytocin (OT) is a pleiotropic hormone produced in the paraventricular and supraoptic nuclei of the hypothalamus and released into the bloodstream through the posterior pituitary gland and into the brain through several pathways [47]. Oxytocin seems to promote social interactions between conspecifics, attachment, and pair-bonding, and it also exhibits anti-inflammatory properties as a powerful immune and stress system modulator, beyond its “classical” activities, such as uterine contractions during labor and milk production [48][49][50] (Figure 3).
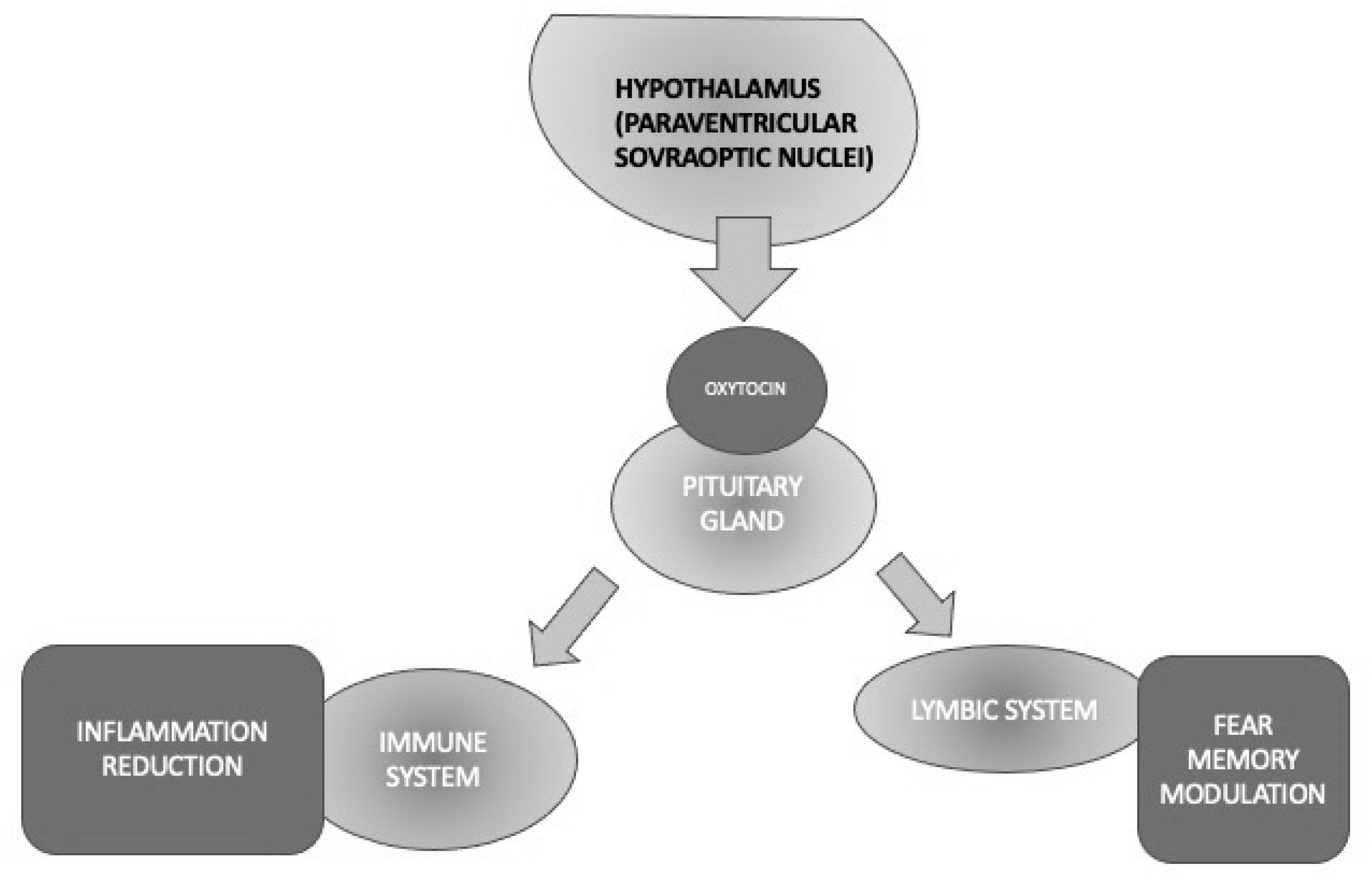
Figure 3. Different activities of oxytocin implicated in PTSD through its anti-inflammatory and fear memory modulation properties.
The first evidence that the OT might be involved in the pathophysiology of PTSD derived from studies on animals showing that exogenous OT injection reduced sympathetic responses, such as heart rate, blood pressure, and cortisol levels, and attenuated the activation of the HPA axis in threatening situations [51][52]. Such findings highlighted that these effects of OT were due to decreased activation of the central amygdala. Data in humans reported a relationship between traumatic events and/or PTSD following severe childhood abuse and reduced endogenous OT concentrations [53][54][55][56]. Genetic association studies revealed that some OTR gene polymorphisms might be associated with an increased risk of experiencing traumatic events and/or developing psychiatric disorders, such as PTSD, anxiety, and depression, while other OTR gene polymorphisms may have a protective role. The most studied polymorphism, namely that of the OXTR rs53576 GG genotype, was associated with insecure attachments, poor response to social support, emotional dysregulation, and less resilience to stress, all factors that are linked to a greater vulnerability to psychiatric disorders related to traumatic experiences [57]. The direct measurements of plasma OT levels in patients with PTSD are few and controversial while reporting controversial findings with women generally reported to show higher concentrations than men [58][59]. The reported differences could be explained by the fact that sex hormones affect OT release and its receptor expression.
To sum up, OT is a hormone that has been well known for decades for exerting the modulation of a variety of functions that are core elements of PTSD, such as stress and fear/anxiety responses. The exact role of OT in stress processes is still unclear; however, it has been reported to decrease the response of the HPA axis in rodents and primates. Other physiological OT activities, including the attenuation of memory consolidation and retrieval, facilitation of the extinction of an activated avoidance response, and decrease of passive avoidance behavior, would support its potential role in PTSD neurobiology. The availability of intranasal OT that has been preliminarily demonstrated to be effective in other psychiatric conditions requires it to be tested in large samples of PTSD, given its relative absence of significant side effects, except for a transient amnesia that would be beneficial just in PTSD. Therefore, OT seems to represent a worthy target to investigate the pathophysiology and/or develop future drugs to treat this condition [60] (Figure 3).
7. Neuropeptide Y
Neuropeptide Y (NPY) is a 36-amino-acid peptide that has widespread physiological and behavioral functions in the CNS and peripheral organs [61]. The physiological functions of NPY include the regulation of feeding behavior, energy homeostasis, blood pressure, reproductive behavior, and memory. Neuropeptide Y plays an important, albeit complex, role in regulating responses to stress and modulating fear and anxiety-related behaviors [61]. The different effects of NPY are mediated by at least five different G-protein-coupled receptors called Y1, Y2, Y4, Y5, and Y6 (Figure 4). Converging preclinical evidence supports NPY’s role in modulating the stress response and regulating anxiety in animal models. Advances in translational research point to NPY as a key mediator of stress response deserving attention for its potential therapeutic activity in PTSD. Therefore, both agonists of the Y2 and Y4 receptors appear to be potential novel treatments for anxiety disorders [62]. The combination of Y1 agonist and Y2 antagonist activity seems to reverse the alterations in learning and memory processes typical of PTSD [61]. All these findings are interesting but still preliminary and hypothetical (Figure 4).
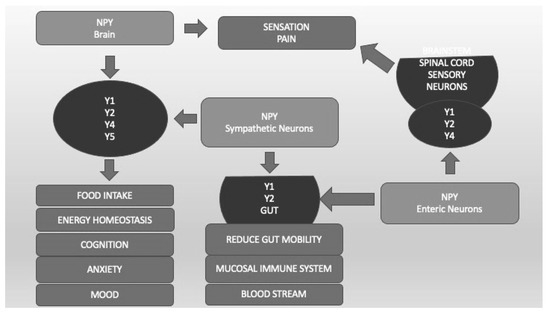
Figure 4. Neuropeptide Y (NPY) and its different receptors (Y1, Y2, Y4, Y5). Y1, Y2, Y4, and Y5, localized in different CNS areas and bound by NPY produced by CNS and sympathetic neurons, are important not only for food intake and energy homeostasis but also for cognition, anxiety, and mood regulation. Sensation and pain are regulated by CNS and enteric neurons that bind Y1, Y2, and Y4 localized in spinal cord sensory neurons. Y1 and Y2, located in the Gut system and bound by CNS and enteric NPY, regulate blood pressure, mucosal immunity, and gut mobility.
8. MicroRNA
The microRNAs (miRNAs) are non-coding, single-stranded RNA molecules genomically encoded with 19–24 nucleotides that are coated with sequences complementary to messenger RNA (mRNA), thus regulating protein expression. Starting from the consideration that PTSD requires exposure to a traumatic event and that genes are sensitive to stress and trauma, epigenetic alterations have received attention as a possible mechanism for the development and persistence of PTSD [63][64][65][66][67]. Epigenetic modifications, including DNA methylation, histone modifications, and ncRNA, have been implicated in a number of complex diseases, such as cardiovascular disease, cancer, and neurological diseases [68]. Abnormalities in miRNA expression can fine-tune the expression of multiple genes within a biological network, suggesting that its dysregulation may be crucial for many of the observed molecular changes in the pathogenesis of PTSD. These findings provide evidence that miRNA not only plays a critical role in the pathophysiology of PTSD but can also open up new avenues for the development of diagnostic tools and therapeutic targets for the PTSD phenotype.
Given the paucity of human trials, animal models simulating pathophysiological features of these disorders, such as deficits in fear extinction, have significantly contributed to the understanding of the underlying processes involved in decreasing dysregulated fear. Future miRNA-based treatments for the management of fear-related illnesses, such as phobias and PTSD, hold the intriguing possibility of targeting such miRNAs [69].
9. Pipeline of Currently Studied Drugs
To date, a large proportion of ongoing trials involve different forms of psychotherapy, often implemented together with pharmacological interventions. Indeed, such an integrative approach is demonstrative of the intrinsic complexity of PTSD, which requires addressing both the psychological and biological facets of the disorder. In addition, there is a growing interest in exploring unconventional and innovative therapeutic modalities that, while being remarkably eclectic, perfectly illustrate the heterogeneity of contemporary research in this field. These range from paired vagus nerve stimulation and neurofeedback to hyperbaric oxygen therapy and translingual neurostimulation. The investigation of eye movement desensitization reprocessing therapy, deep brain stimulation (DBS), and equine assisted therapy (EAT) further exemplifies the breadth and depth of current research efforts. Remarkably, the scope of investigation extends beyond the conventional boundaries of medicine and into holistic practices, as exemplified by trials studying the effects of Sudarshan Kriya Yoga (SKY) in PTSD management. This intriguing blend of conventional and complementary approaches signifies a paradigm shift in the understanding and management of PTSD.
As a matter of fact, the trajectory of pharmacotherapeutic research for PTSD will further expand to include an array of new potential medications, as seen in the most recent clinical trials. For instance, the NCT05401565 study is evaluating the therapeutic potential of balovaptan, a vasopressin 1a receptor antagonist. Of note, another distinctive investigation involves the antiviral combination of glecaprevir and pibrentasvir for PTSD treatment [NCT05446857]. Specifically, this study aims at examining the effects of these hepatitis C virus-targeting agents on PTSD, hypothesizing that the therapeutic action might extend beyond their original antiviral intent. The results may potentially expand the understanding of the psychoneuroimmunological factors at play in PTSD, offering novel avenues for therapeutic intervention.
Finally, the boundaries of PTSD pharmacotherapy are being pushed even further with the investigation of methylone [NCT05741710], a psychoactive substance structurally related to the ‘club drug’ MDMA, building upon previous findings that highlight the therapeutic potential of similar substances. This groundbreaking research could redefine the perceptions of the therapeutic utility of such psychoactive substances in the management of PTSD and other mental health disorders.
Undeniably, the investigations identified through the comprehensive exploration of ongoing clinical trials reflect a holistic and innovative approach to combating PTSD. Firstly, it is crucial to note that these trials represent the current forefront of PTSD pharmacotherapy research. From the repurposing of drugs such as doxazosin, clonidine, and dronabinol to the exploration of novel compounds like BNC210, BI 1358894, JZP150, balovaptan, glecaprevir/pibrentasvir, and methylone, the PTSD pharmacotherapy pipeline is undeniably rich and diverse. Secondly, the exploration of different treatment modalities such as the intravenous administration of brexanolone and the use of stellate ganglion block with bupivacaine highlights the tireless pursuit of optimal treatment strategies to enhance the clinical management of PTSD.
A conspicuous aspect of the ongoing investigation remains the emphasis on nightmares and sleep disturbances. These are prominent symptoms of PTSD, and their repeated appearance in these trials underscores the importance of addressing these symptoms for comprehensive PTSD management. Indeed, this collective focus further reinforces the significance of a multi-modal treatment approach, recognizing the complex symptomatology of PTSD that extends beyond mere psychological manifestations. In the context of a global drug development perspective, these trials represent a concerted global effort in the quest for more effective pharmacological interventions for PTSD. Not only do they embody a dynamic and global endeavor to improve the lives of those living with PTSD, but they also serve to bridge the gap between the understanding of the mechanistic underpinnings of PTSD and the development of efficacious, target-oriented therapeutic strategies.
This entry is adapted from the peer-reviewed paper 10.3390/life13081731
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision DSM-5-TR, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2022.
- Barbara Young Welke. Recasting American Liberty: Gender, Race, Law, and the Railroad Revolution, 1865–1920; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2001.
- Birmes, P.; Hatton, L.; Brunet, A.; Schmitt, L. Early historical literature for post-traumatic symptomatology. Stress Health 2003, 19, 17–26.
- Jones, E. Historical approaches to post-combat disorders. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 533–542.
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-traumatic stress disorder. Nat. Rev. Dis. Primers 2015, 1, 15057.
- Kessler, R.C. Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 1995, 52, 1048–1060.
- Brewin, C.R.; Andrews, B.; Valentine, J.D. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J. Consult. Clin. Psychol. 2000, 68, 748–766.
- Tull, M.T.; Forbes, C.N.; Weiss, N.H.; Gratz, K.L. An investigation of the effect of trauma script exposure on risk-taking among patients with substance use disorders and posttraumatic stress disorder. J. Anxiety Disord. 2019, 62, 77–85.
- Sundin, J.; Herrell, R.K.; Hoge, C.W.; Fear, N.T.; Adler, A.B.; Greenberg, N.; Riviere, L.A.; Thomas, J.L.; Wessely, S.; Bliese, P.D. Mental health outcomes in US and UK military personnel returning from Iraq. Br. J. Psychiatry 2014, 204, 200–207.
- Lok, A.; Frijling, J.L.; van Zuiden, M. Posttraumatische stressstoornis . Ned Tijdschr Geneeskd. 2018, 161, D1905. (In Dutch)
- Fox, V.; Dalman, C.; Dal, H.; Hollander, A.-C.; Kirkbride, J.B.; Pitman, A. Suicide risk in people with post-traumatic stress disorder: A cohort study of 3.1 million people in Sweden. J. Affect. Disord. 2020, 279, 609–616.
- Fink, D.S.; Gradus, J.L.; Keyes, K.M.; Calabrese, J.R.; Liberzon, I.; Tamburrino, M.B.; Cohen, G.H.; Sampson, L.; Galea, S. Subthreshold PTSD and PTSD in a prospective-longitudinal cohort of military personnel: Potential targets for preventive interventions. Depress. Anxiety 2018, 35, 1048–1055.
- Ressler, K.J.; Berretta, S.; Bolshakov, V.Y.; Rosso, I.M.; Meloni, E.G.; Rauch, S.L.; Carlezon, W.A. Post-traumatic stress disorder: Clinical and translational neuroscience from cells to circuits. Nat. Rev. Neurol. 2022, 18, 273–288.
- de Silva, V.A.; Jayasekera, N.; Hanwella, R. Cannabis Use among Navy personnel in Sri Lanka: A cross sectional study. BMC Res. Notes 2016, 9, 174.
- Kelmendi, B.; Adams, T.G.; Yarnell, S.; Southwick, S.; Abdallah, C.G.; Krystal, J.H. PTSD: From neurobiology to pharmacological treatments. Eur. J. Psychotraumatol. 2016, 7, 31858.
- Snijders, C.; de Nijs, L.; Baker, D.G.; Hauger, R.L.; Hove, D.v.D.; Kenis, G.; Nievergelt, C.M.; Boks, M.P.; Vermetten, E.; Gage, F.H.; et al. MicroRNAs in post-traumatic stress disorder. Behav. Neurobiol. PTSD 2017, 38, 23–46.
- Dunlop, B.W.; Wong, A. The hypothalamic-pituitary-adrenal axis in PTSD: Pathophysiology and treatment interventions. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 361–379.
- Nikbakhtzadeh, M.; Borzadaran, F.M.; Zamani, E.; Shabani, M. Protagonist role of opioidergic system on post-traumatic stress disorder and associated pain. Psychiatry Investig. 2020, 17, 506–516.
- Heinrichs, S.C.; Lapsansky, J.; Lovenberg, T.W.; De Souza, E.B.; Chalmers, D.T. Corticotropin-releasing factor CRF1, but not CRF2, receptors mediate anxiogenic-like behavior. Regul. Pept. 1997, 71, 15–21.
- Cain, C.K.; Maynard, G.D.; Kehne, J.H. Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin. Investig. Drugs 2012, 21, 1323–1350.
- Juruena, M.F. Early-Life Stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav. 2014, 38, 148–159.
- Juruena, M.F.; Eror, F.; Cleare, A.J.; Young, A.H. The role of early life stress in HPA axis and anxiety. Adv. Exp. Med. Biol. 2020, 1191, 141–153.
- Murphy, F.; Nasa, A.; Cullinane, D.; Raajakesary, K.; Gazzaz, A.; Sooknarine, V.; Haines, M.; Roman, E.; Kelly, L.; O’Neill, A.; et al. Childhood trauma, the HPA axis and psychiatric illnesses: A targeted literature synthesis. Front. Psychiatry 2022, 13, 748372.
- Hauger, R.L.; Olivares-Reyes, J.A.; Dautzenberg, F.M.; Lohr, J.B.; Braun, S.; Oakley, R.H. Molecular and cell signaling targets for ptsd pathophysiology and pharmacotherapy. Neuropharmacology 2012, 62, 705–714.
- Shang, Y.; Filizola, M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol. 2015, 763, 206–213.
- Emery, M.A.; Eitan, S. Members of the same pharmacological family are not alike: Different opioids, different consequences, hope for the opioid crisis? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 428–449.
- Moore, K.A.; Werner, C.; Zannelli, R.M.; Levine, B.; Smith, M.L. Screening postmortem blood and tissues for nine cases of drugs of abuse using automated microplate immunoassay. Forensic Sci. Int. 1999, 106, 93–102.
- Adem, A.; Madjid, N.; Kahl, U.; Holst, S.; Sadek, B.; Sandin, J.; Terenius, L.; Ögren, S.O. Nociceptin and the NOP receptor in aversive learning in mice. Eur. Neuropsychopharmacol. 2017, 27, 1298–1307.
- Saxe, G.; Stoddard, F.; Courtney, D.; Cunningham, K.; Chawla, N.; Sheridan, R.; King, D.; King, L. Relationship between acute morphine and the course of PTSD in children with burns. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 915–921.
- Bryant, R.A.; Creamer, M.; O’Donnell, M.; Silove, D.; McFarlane, A.C. A study of the protective function of acute morphine administration on subsequent posttraumatic stress disorder. Biol. Psychiatry 2009, 65, 438–440.
- Likhtik, E.; Popa, D.; Apergis-Schoute, J.; Fidacaro, G.A.; Paré, D. Amygdala intercalated neurons are required for expression of fear extinction. Nature 2008, 454, 642–645.
- Seal, K.H.; Shi, Y.; Cohen, G.; Cohen, B.E.; Maguen, S.; Krebs, E.E.; Neylan, T.C. Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA 2012, 307, 940–947.
- Shiner, B.; Leonard Westgate, C.; Bernardy, N.C.; Schnurr, P.P.; Watts, B.V. Trends in opioid use disorder diagnoses and medication treatment among veterans with posttraumatic stress disorder. J. Dual Diagn. 2017, 13, 201–212.
- Kelley, A.T.; Greenstone, C.L.; Kirsh, S.R. Defining access and the role of community care in the veterans health administration. J. Gen. Intern. Med. 2019, 35, 1584–1585.
- Stoddard, F.J.; Sorrentino, E.A.; Ceranoglu, T.A.; Saxe, G.; Murphy, J.M.; Drake, J.E.; Ronfeldt, H.; White, G.W.; Kagan, J.; Snidman, N.; et al. Preliminary evidence for the effects of morphine on posttraumatic stress disorder symptoms in one- to four-year-olds with burns. J. Burn Care Res. 2009, 30, 836–843.
- Norman, S.B.; Inaba, R.K.; Smith, T.L.; Brown, S.A. Development of the PTSD-Alcohol Expectancy Questionnaire. Addict. Behav. 2008, 33, 841–847.
- Porto, G.P.; Milanesi, L.H.; Rubin, M.A.; Mello, C.F. Effect of morphine on the persistence of long-term memory in rats. Psychopharmacology 2014, 232, 1747–1753.
- Madison, C.A.; Eitan, S. Buprenorphine: Prospective novel therapy for depression and PTSD. Psychol. Med. 2020, 50, 881–893.
- Nedergaard, M.; Takano, T.; Hansen, A.J. Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 2002, 3, 748–755.
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 298–487.
- Riedel, G. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47.
- Peng, S.; Zhang, Y.; Zhang, J.; Wang, H.; Ren, B. Glutamate receptors and signal transduction in learning and memory. Mol. Biol. Rep. 2010, 38, 453–460.
- Vargas, A.S.; Luís, Â.; Barroso, M.; Gallardo, E.; Pereira, L. Psilocybin as a New Approach to Treat Depression and Anxiety in the Context of Life-Threatening Diseases-A Systematic Review and Meta-Analysis of Clinical Trials. Biomedicines 2020, 8, 331.
- Zanikov, T.; Gerasymchuk, M.; Ghasemi Gojani, E.; Robinson, G.I.; Asghari, S.; Groves, A.; Haselhorst, L.; Nandakumar, S.; Stahl, C.; Cameron, M.; et al. The Effect of Combined Treatment of Psilocybin and Eugenol on Lipopolysaccharide-Induced Brain Inflammation in Mice. Molecules 2023, 28, 2624.
- Davis, A.K.; Levin, A.W.; Nagib, P.B.; Armstrong, S.B.; Lancelotta, R.L. Study Protocol of an Open-Label Proof-of-Concept Trial Examining the Safety and Clinical Efficacy of Psilocybin-Assisted Therapy for Veterans with PTSD. BMJ Open 2023, 13, e068884.
- Lu, H.-C.; Mackie, K. An Introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525.
- Bolsoni, L.M.; Crippa, J.A.S.; Hallak, J.E.C.; Guimarães, F.S.; Zuardi, A.W. Effects of cannabidiol on symptoms induced by the recall of traumatic events in patients with posttraumatic stress disorder. Psychopharmacology 2022, 239, 1499–1507.
- Carbone, M.G.; Marazziti, D.; Diep, P.-T.; Carter, S. Oxytocin: An old hormone, a novel psychotropic drug and its possible use in treating psychiatric disorders. Curr. Med. Chem. 2022, 29, 5615–5687.
- Carter, C.S. Oxytocin pathways and the evolution of human behavior. Annu. Rev. Psychol. 2014, 65, 17–39.
- Marsh, N.; Marsh, A.A.; Lee, M.R.; Hurlemann, R. Oxytocin and the neurobiology of prosocial behavior. Neuroscientist 2020, 27, 604–619.
- Petersson, M.; Hulting, A.-L.; Uvnäs-Moberg, K. oxytocin causes a sustained decrease in plasma levels of corticosterone in rats. Neurosci. Lett. 1999, 264, 41–44.
- Petersson, M.; Uvnäs-Moberg, K. Systemic oxytocin treatment modulates glucocorticoid and mineralocorticoid receptor mRNA in the rat hippocampus. Neurosci. Lett. 2003, 343, 97–100.
- Heim, C.; Young, L.J.; Newport, D.J.; Mletzko, T.; Miller, A.H.; Nemeroff, C.B. Lower CSF oxytocin concentrations in women with a history of childhood abuse. Mol. Psychiatry 2008, 14, 954–958.
- Chatzittofis, A.; Nordström, P.; Uvnäs-Moberg, K.; Asberg, M.; Jokinen, J. CSF and plasma oxytocin levels in suicide attempters, the role of childhood trauma and revictimization. Neuro Endocrinol. Lett. 2014, 35, 213–217.
- Mohiyeddini, C.; Opacka-Juffry, J.; Gross, J.J. Emotional suppression explains the link between early life stress and plasma oxytocin. Anxiety Stress Coping 2014, 27, 466–475.
- Mizuki, R.; Fujiwara, T. Association of oxytocin level and less severe forms of childhood maltreatment history among healthy Japanese adults involved with child care. Front. Behav. Neurosci. 2015, 9, 138.
- Carmassi, C.; Marazziti, D.; Mucci, F.; Della Vecchia, A.; Barberi, F.M.; Baroni, S.; Giannaccini, G.; Palego, L.; Massimetti, G.; Dell’osso, L. Decreased plasma oxytocin levels in patients with PTSD. Front. Psychol. 2021, 12, 612338.
- Marazziti, D.; Baroni, S.; Mucci, F.; Piccinni, A.; Moroni, I.; Giannaccini, G.; Carmassi, C.; Massimetti, E.; Dell’osso, L. Sex-related differences in plasma oxytocin levels in humans. Clin. Pract. Epidemiol. Ment. Health 2019, 15, 58–63.
- Frijling, J.L.; van Zuiden, M.; Nawijn, L.; Koch, S.B.J.; Neumann, I.D.; Veltman, D.J.; Olff, M. Salivary oxytocin and vasopressin levels in police officers with and without post-traumatic stress disorder. J. Neuroendocrinol. 2015, 27, 743–751.
- Olff, M.; Langeland, W.; Witteveen, A.; Denys, D. A psychobiological rationale for oxytocin in the treatment of posttraumatic stress disorder. CNS Spectr. 2010, 15, 522–530.
- Kautz, M.; Charney, D.S.; Murrough, J.W. Neuropeptide Y, resilience, and PTSD therapeutics. Neurosci. Lett. 2017, 649, 164–169.
- Tasan, R.; Verma, D.; Wood, J.; Lach, G.; Hörmer, B.; de Lima, T.; Herzog, H.; Sperk, G. The role of neuropeptide y in fear conditioning and extinction. Neuropeptides 2016, 55, 111–126.
- Roth, T.L.; Zoladz, P.R.; Sweatt, J.D.; Diamond, D.M. Epigenetic modification of hippocampal BDNF DNA in adult rats in an animal model of post-traumatic stress disorder. J. Psychiatr. Res. 2011, 45, 919–926.
- Mehta, D.; Bruenig, D.; Carrillo-Roa, T.; Lawford, B.; Harvey, W.; Morris, C.P.; Smith, A.K.; Binder, E.B.; Young, R.M.; Voisey, J. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr. Scand. 2017, 136, 493–505.
- Rutten, B.P.F.; Vermetten, E.; Vinkers, C.H.; Ursini, G.; Daskalakis, N.P.; Pishva, E.; De Nijs, L.; Houtepen, L.C.; Eijssen, L.; Jaffe, A.E.; et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol. Psychiatry 2017, 23, 1145–1156.
- Al Jowf, G.I.; Snijders, C.; Rutten, B.P.F.; de Nijs, L.; Eijssen, L.M.T. The molecular biology of susceptibility to post-traumatic stress disorder: Highlights of epigenetics and epigenomics. Int. J. Mol. Sci. 2021, 22, 10743.
- Maurel, O.M.; Torrisi, S.A.; Barbagallo, C.; Purrello, M.; Salomone, S.; Drago, F.; Ragusa, M.; Leggio, G.M. Dysregulation of MiR-15a-5p, MiR-497a-5p and MiR-511-5p is associated with modulation of BDNF and FKBP5 in brain areas of PTSD-related susceptible and resilient mice. Int. J. Mol. Sci. 2021, 22, 5157.
- Klengel, T.; Binder, E.B. Allele-specific epigenetic modification: A molecular mechanism for gene–environment interactions in stress-related psychiatric disorders? Epigenomics 2013, 5, 109–112.
- Murphy, C.P.; Singewald, N. Potential of microRNAs as novel targets in the alleviation of pathological fear. Genes Brain Behav. 2018, 17, e12427.
This entry is offline, you can click here to edit this entry!