HIV-1 has evolved a plethora of strategies to overcome the cytoskeletal barrier (i.e., actin and intermediate filaments (AFs and IFs) and microtubules (MTs)) to achieve the viral cycle. HIV-1 modifies cytoskeletal organization and dynamics by acting on associated adaptors and molecular motors to productively fuse, enter, and infect cells and then traffic to the cell surface, where virions assemble and are released to spread infection. The HIV-1 envelope (Env) initiates the cycle by binding to and signaling through its main cell surface receptors (CD4/CCR5/CXCR4) to shape the cytoskeleton for fusion pore formation, which permits viral core entry. Then, the HIV-1 capsid is transported to the nucleus associated with cytoskeleton tracks under the control of specific adaptors/molecular motors, as well as HIV-1 accessory proteins. Furthermore, HIV-1 drives the late stages of the viral cycle by regulating cytoskeleton dynamics to assure viral Pr55Gag expression and transport to the cell surface, where it assembles and buds to mature infectious virions.
- HIV-1 infection
- HIV-1 Env complex
- HIV-1 Pr55Gag
- cytoskeleton dynamics
- cytoskeleton adaptors and motor proteins
1. Actin Filaments
| Cell Factor | Function of the Cellular Factor | Impact on HIV-1 Infection 1 |
|---|---|---|
| ERM | Secure efficient virus spread not only by enhancing virion infectivity but also by preventing excessive membrane fusion at the virological synapse (VS). | + |
| EWI-2/CD81 | Difficult HIV-1 Env-mediates cell-to-cell fusion. | − |
| BST2/tetherin | Links AFs with plasma membrane where it sequesters the viral Env and preventing the virus escape from cell. | − |
| PSGL-1 | Inhibits RT in target cells and diminished the infectivity of released virions. | − |
| Arp2/3 and Rac-1/Cdc42- IQGAP1 pathways | Remodeling of cortical AFs in areas of high positive membrane curvature, within Arp2/3- and Rac1/Cdc42/IQGAP1-dependent F-actin filopodia and lamellipodia structures, which enables HIV-1 to bud and cell-to-cell spreading. | + |
| AFs | Cortical AFs bind HIV-1 Pr55Gag protein by the NC domain. | + |
| Viral factor | Function of the viral factor | Impact on HIV-1 infection 1 |
| Pr55Gag | Actin interaction facilitating viral egress. | + |
| Vpu | Counteracts BST-2/tetherin, EWI-2, and PSGL-1 antiviral activities. | + |
2. Microtubules
| Cell Factor | Function of the Cellular Factor | Impact on HIV-1 Infection 1 |
|---|---|---|
| HDAC6 | Colocalized with and deacetylates MTs to promote autophagy degradation of Vif and Pr55Gag viral proteins, thus stabilizing the restriction factor A3G, forming an A3G/HDAC6 antiviral complex, an inhibiting HIV-1 viral production and infectiveness. | − |
| TDP-43 | Regulates HDAC6 mRNA and protein levels promoting its antiviral function by tubulin-deacetylation and dependent anti-HIV-1 autophagy. Controls HIV-1 production and the infection capacity of viral particles. | − |
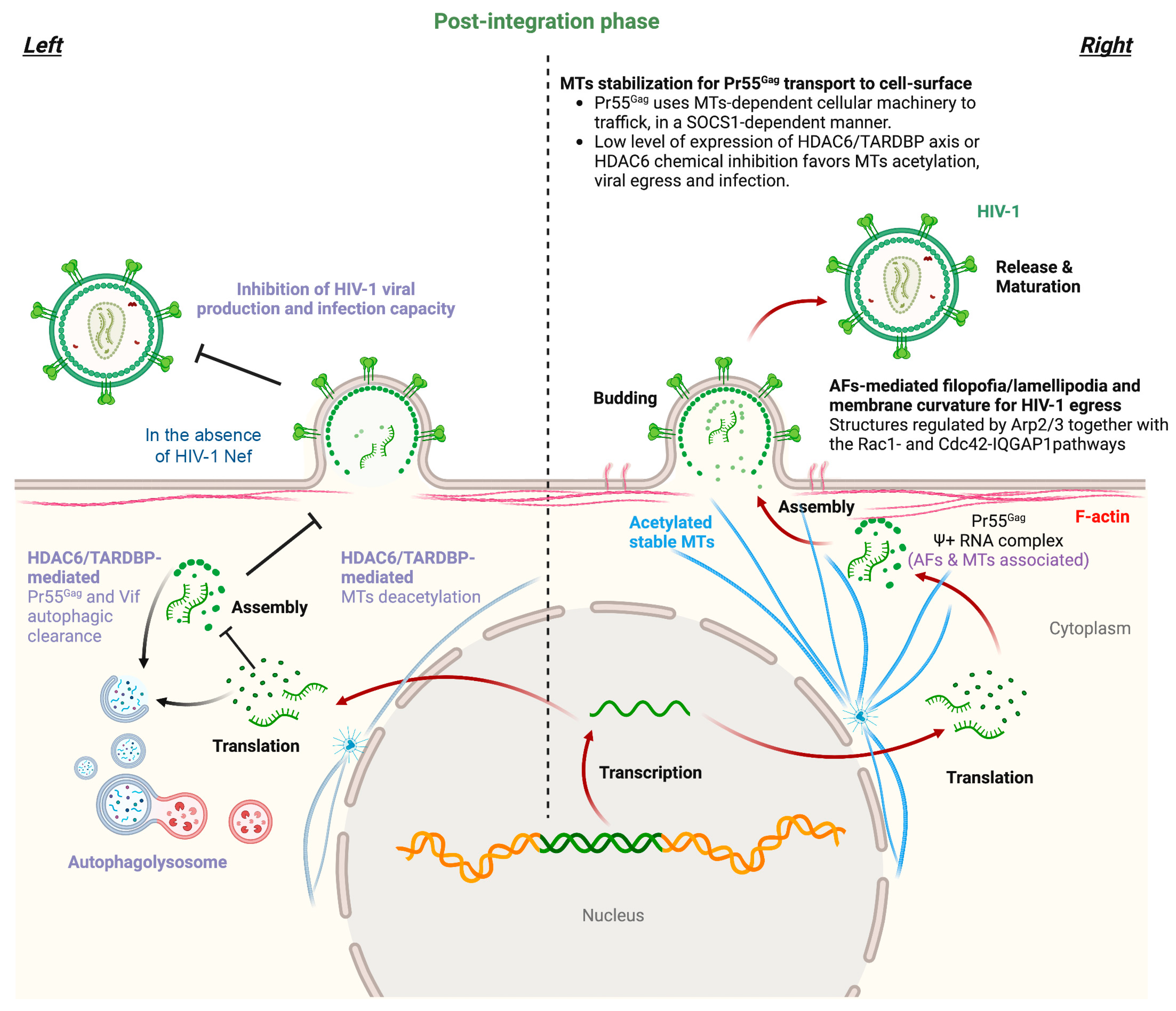
| SOCS1 | Associates with MTs and allows Pr55Gag efficient transport to plasma membrane. | + |
| IQGAP1 | A MTs-associated signaling scaffold protein that impedes viral egress by interacting with HIV-1 Pr55Gag, affecting its distribution and expression at plasma membrane-budding areas. | − |
| Viral factor | Function of the viral factor | Impact on HIV-1 infection 1, cell-function or viral toxicity 2 |
| Nef | Targets tubulin-deacetylase HDAC6 to stabilize MTs, HIV-1 Pr55Gag and Vif proteins, assuring virus production and the infection capacity of HIV-1. | + |
| Pr55Gag | HIV-1 Pr55Gag uses MTs-dependent cellular machinery to traffic to the cell-surface in a SOCS1-dependent manner. | + |
| Tat | Interacts with tubulin leading to the alteration of MTs dynamics and the activation of a mitochondria-dependent apoptotic pathway. Tat uses Bim to facilitate this MTs-associated cell-death signal. | * |
| Vpr | Modulator of the MTs-dependent endocytic trafficking, negatively affecting phagosome biogenesis and maturation. | * |
3. Intermediate Filaments
This entry is adapted from the peer-reviewed paper 10.3390/ijms241713104
References
- Puigdomenech, I.; Massanella, M.; Cabrera, C.; Clotet, B.; Blanco, J. On the steps of cell-to-cell HIV transmission between CD4 T cells. Retrovirology 2009, 6, 89.
- De Armas-Rillo, L.; Valera, M.S.; Marrero-Hernandez, S.; Valenzuela-Fernandez, A. Membrane dynamics associated with viral infection. Rev. Med. Virol. 2016, 26, 146–160.
- Bracq, L.; Xie, M.; Benichou, S.; Bouchet, J. Mechanisms for Cell-to-Cell Transmission of HIV-1. Front. Immunol. 2018, 9, 260.
- Barroso-Gonzalez, J.; Garcia-Exposito, L.; Puigdomenech, I.; de Armas-Rillo, L.; Machado, J.D.; Blanco, J.; Valenzuela-Fernandez, A. Viral infection: Moving through complex and dynamic cell-membrane structures. Commun. Integr. Biol. 2011, 4, 398–408.
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Kohler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219.
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315.
- Do, T.; Murphy, G.; Earl, L.A.; Del Prete, G.Q.; Grandinetti, G.; Li, G.H.; Estes, J.D.; Rao, P.; Trubey, C.M.; Thomas, J.; et al. Three-dimensional imaging of HIV-1 virological synapses reveals membrane architectures involved in virus transmission. J. Virol. 2014, 88, 10327–10339.
- Hashimoto, M.; Bhuyan, F.; Hiyoshi, M.; Noyori, O.; Nasser, H.; Miyazaki, M.; Saito, T.; Kondoh, Y.; Osada, H.; Kimura, S.; et al. Potential Role of the Formation of Tunneling Nanotubes in HIV-1 Spread in Macrophages. J. Immunol. 2016, 196, 1832–1841.
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660.
- Eugenin, E.A.; Gaskill, P.J.; Berman, J.W. Tunneling nanotubes (TNT): A potential mechanism for intercellular HIV trafficking. Commun. Integr. Biol. 2009, 2, 243–244.
- Eugenin, E.A.; Gaskill, P.J.; Berman, J.W. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell Immunol. 2009, 254, 142–148.
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325.
- Sutton, J.; Dotson, D.; Dong, X. Molecular Events in Late Stages of HIV-1 Replication. JSM Microbiol. 2013, 1, 1004.
- Aggarwal, A.; Iemma, T.L.; Shih, I.; Newsome, T.P.; McAllery, S.; Cunningham, A.L.; Turville, S.G. Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells. PLoS Pathog. 2012, 8, e1002762.
- Ljubojevic, N.; Henderson, J.M.; Zurzolo, C. The Ways of Actin: Why Tunneling Nanotubes Are Unique Cell Protrusions. Trends Cell Biol. 2021, 31, 130–142.
- Roy, N.H.; Lambele, M.; Chan, J.; Symeonides, M.; Thali, M. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. J. Virol. 2014, 88, 7645–7658.
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113.
- Whitaker, E.E.; Matheson, N.J.; Perlee, S.; Munson, P.B.; Symeonides, M.; Thali, M. EWI-2 Inhibits Cell-Cell Fusion at the HIV-1 Virological Presynapse. Viruses 2019, 11, 1082.
- Andreau, K.; Perfettini, J.L.; Castedo, M.; Metivier, D.; Scott, V.; Pierron, G.; Kroemer, G. Contagious apoptosis facilitated by the HIV-1 envelope: Fusion-induced cell-to-cell transmission of a lethal signal. J. Cell Sci. 2004, 117, 5643–5653.
- Ferri, K.F.; Jacotot, E.; Geuskens, M.; Kroemer, G. Apoptosis and karyogamy in syncytia induced by the HIV-1-envelope glycoprotein complex. Cell Death Differ. 2000, 7, 1137–1139.
- Nardacci, R.; Perfettini, J.L.; Grieco, L.; Thieffry, D.; Kroemer, G.; Piacentini, M. Syncytial apoptosis signaling network induced by the HIV-1 envelope glycoprotein complex: An overview. Cell Death Dis. 2015, 6, e1846.
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006, 177, 3260–3265.
- Ishikawa, J.; Kaisho, T.; Tomizawa, H.; Lee, B.O.; Kobune, Y.; Inazawa, J.; Oritani, K.; Itoh, M.; Ochi, T.; Ishihara, K.; et al. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 1995, 26, 527–534.
- Rollason, R.; Korolchuk, V.; Hamilton, C.; Jepson, M.; Banting, G. A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J. Cell Biol. 2009, 184, 721–736.
- Fitzpatrick, K.; Skasko, M.; Deerinck, T.J.; Crum, J.; Ellisman, M.H.; Guatelli, J. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 2010, 6, e1000701.
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511.
- Hammonds, J.; Wang, J.J.; Yi, H.; Spearman, P. Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010, 6, e1000749.
- Habermann, A.; Krijnse-Locker, J.; Oberwinkler, H.; Eckhardt, M.; Homann, S.; Andrew, A.; Strebel, K.; Krausslich, H.G. CD317/tetherin is enriched in the HIV-1 envelope and downregulated from the plasma membrane upon virus infection. J. Virol. 2010, 84, 4646–4658.
- Hammonds, J.; Wang, J.J.; Spearman, P. Restriction of Retroviral Replication by Tetherin/BST-2. Mol. Biol. Int. 2012, 2012, 424768.
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483.
- Giese, S.; Marsh, M. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog. 2014, 10, e1004189.
- Kuhl, B.D.; Sloan, R.D.; Donahue, D.A.; Bar-Magen, T.; Liang, C.; Wainberg, M.A. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 2010, 7, 115.
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430.
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252.
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Mercier, J.; Binette, J.; Leung, G.; Cohen, E.A. Suppression of Tetherin-restricting activity upon human immunodeficiency virus type 1 particle release correlates with localization of Vpu in the trans-Golgi network. J. Virol. 2009, 83, 4574–4590.
- Varthakavi, V.; Smith, R.M.; Martin, K.L.; Derdowski, A.; Lapierre, L.A.; Goldenring, J.R.; Spearman, P. The pericentriolar recycling endosome plays a key role in Vpu-mediated enhancement of HIV-1 particle release. Traffic 2006, 7, 298–307.
- Schubert, U.; Strebel, K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 1994, 68, 2260–2271.
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a betaTrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947.
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297.
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072.
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009, 5, e1000574.
- Skasko, M.; Wang, Y.; Tian, Y.; Tokarev, A.; Munguia, J.; Ruiz, A.; Stephens, E.B.; Opella, S.J.; Guatelli, J. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 2012, 287, 58–67.
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450.
- Liu, Y.; Song, Y.; Zhang, S.; Diao, M.; Huang, S.; Li, S.; Tan, X. PSGL-1 inhibits HIV-1 infection by restricting actin dynamics and sequestering HIV envelope proteins. Cell Discov. 2020, 6, 53.
- Goetz, D.J.; Greif, D.M.; Ding, H.; Camphausen, R.T.; Howes, S.; Comess, K.M.; Snapp, K.R.; Kansas, G.S.; Luscinskas, F.W. Isolated P-selectin glycoprotein ligand-1 dynamic adhesion to P- and E-selectin. J. Cell Biol. 1997, 137, 509–519.
- Xia, L.; Ramachandran, V.; McDaniel, J.M.; Nguyen, K.N.; Cummings, R.D.; McEver, R.P. N-terminal residues in murine P-selectin glycoprotein ligand-1 required for binding to murine P-selectin. Blood 2003, 101, 552–559.
- Spertini, O.; Cordey, A.S.; Monai, N.; Giuffre, L.; Schapira, M. P-selectin glycoprotein ligand 1 is a ligand for L-selectin on neutrophils, monocytes, and CD34+ hematopoietic progenitor cells. J. Cell Biol. 1996, 135, 523–531.
- Baisse, B.; Galisson, F.; Giraud, S.; Schapira, M.; Spertini, O. Evolutionary conservation of P-selectin glycoprotein ligand-1 primary structure and function. BMC Evol. Biol. 2007, 7, 166.
- Carlow, D.A.; Gossens, K.; Naus, S.; Veerman, K.M.; Seo, W.; Ziltener, H.J. PSGL-1 function in immunity and steady state homeostasis. Immunol. Rev. 2009, 230, 75–96.
- Moore, K.L. Structure and function of P-selectin glycoprotein ligand-1. Leuk Lymphoma 1998, 29, 1–15.
- Liu, Y.; Fu, Y.; Wang, Q.; Li, M.; Zhou, Z.; Dabbagh, D.; Fu, C.; Zhang, H.; Li, S.; Zhang, T.; et al. Proteomic profiling of HIV-1 infection of human CD4(+) T cells identifies PSGL-1 as an HIV restriction factor. Nat. Microbiol. 2019, 4, 813–825.
- Grover, J.R.; Veatch, S.L.; Ono, A. Basic motifs target PSGL-1, CD43, and CD44 to plasma membrane sites where HIV-1 assembles. J. Virol. 2015, 89, 454–467.
- Fu, Y.; He, S.; Waheed, A.A.; Dabbagh, D.; Zhou, Z.; Trinite, B.; Wang, Z.; Yu, J.; Wang, D.; Li, F.; et al. PSGL-1 restricts HIV-1 infectivity by blocking virus particle attachment to target cells. Proc. Natl. Acad. Sci. USA 2020, 117, 9537–9545.
- Aggarwal, A.; Stella, A.O.; Henry, C.C.; Narayan, K.; Turville, S.G. Embedding of HIV Egress within Cortical F-Actin. Pathogens 2022, 11, 56.
- Weissbach, L.; Settleman, J.; Kalady, M.F.; Snijders, A.J.; Murthy, A.E.; Yan, Y.X.; Bernards, A. Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 1994, 269, 20517–20521.
- Bashour, A.M.; Fullerton, A.T.; Hart, M.J.; Bloom, G.S. IQGAP1, a Rac- and Cdc42-binding protein, directly binds and cross-links microfilaments. J. Cell Biol. 1997, 137, 1555–1566.
- Liu, B.; Dai, R.; Tian, C.J.; Dawson, L.; Gorelick, R.; Yu, X.F. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 1999, 73, 2901–2908.
- Poole, E.; Strappe, P.; Mok, H.P.; Hicks, R.; Lever, A.M. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic 2005, 6, 741–755.
- Gladnikoff, M.; Shimoni, E.; Gov, N.S.; Rousso, I. Retroviral assembly and budding occur through an actin-driven mechanism. Biophys. J. 2009, 97, 2419–2428.
- Carlson, L.A.; de Marco, A.; Oberwinkler, H.; Habermann, A.; Briggs, J.A.; Krausslich, H.G.; Grunewald, K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010, 6, e1001173.
- Stauffer, S.; Rahman, S.A.; de Marco, A.; Carlson, L.A.; Glass, B.; Oberwinkler, H.; Herold, N.; Briggs, J.A.; Muller, B.; Grunewald, K.; et al. The nucleocapsid domain of Gag is dispensable for actin incorporation into HIV-1 and for association of viral budding sites with cortical F-actin. J. Virol. 2014, 88, 7893–7903.
- Rahman, S.A.; Koch, P.; Weichsel, J.; Godinez, W.J.; Schwarz, U.; Rohr, K.; Lamb, D.C.; Krausslich, H.G.; Muller, B. Investigating the role of F-actin in human immunodeficiency virus assembly by live-cell microscopy. J. Virol. 2014, 88, 7904–7914.
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol. Biol. Cell 2005, 16, 5445–5454.
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297.
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826.
- Vasiliver-Shamis, G.; Cho, M.W.; Hioe, C.E.; Dustin, M.L. Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J. Virol. 2009, 83, 11341–11355.
- Sol-Foulon, N.; Sourisseau, M.; Porrot, F.; Thoulouze, M.I.; Trouillet, C.; Nobile, C.; Blanchet, F.; di Bartolo, V.; Noraz, N.; Taylor, N.; et al. ZAP-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J. 2007, 26, 516–526.
- Cabrera-Rodriguez, R.; Perez-Yanes, S.; Lorenzo-Sanchez, I.; Estevez-Herrera, J.; Garcia-Luis, J.; Trujillo-Gonzalez, R.; Valenzuela-Fernandez, A. TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. Int. J. Mol. Sci. 2023, 24, 7658.
- Marrero-Hernandez, S.; Marquez-Arce, D.; Cabrera-Rodriguez, R.; Estevez-Herrera, J.; Perez-Yanes, S.; Barroso-Gonzalez, J.; Madrid, R.; Machado, J.D.; Blanco, J.; Valenzuela-Fernandez, A. HIV-1 Nef Targets HDAC6 to Assure Viral Production and Virus Infection. Front. Microbiol. 2019, 10, 2437.
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53.
- Nishi, M.; Ryo, A.; Tsurutani, N.; Ohba, K.; Sawasaki, T.; Morishita, R.; Perrem, K.; Aoki, I.; Morikawa, Y.; Yamamoto, N. Requirement for microtubule integrity in the SOCS1-mediated intracellular dynamics of HIV-1 Gag. FEBS Lett. 2009, 583, 1243–1250.
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465.
- Ryo, A.; Tsurutani, N.; Ohba, K.; Kimura, R.; Komano, J.; Nishi, M.; Soeda, H.; Hattori, S.; Perrem, K.; Yamamoto, M.; et al. SOCS1 is an inducible host factor during HIV-1 infection and regulates the intracellular trafficking and stability of HIV-1 Gag. Proc. Natl. Acad. Sci. USA 2008, 105, 294–299.
- Briggs, M.W.; Sacks, D.B. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003, 4, 571–574.
- Choi, S.; Anderson, R.A. IQGAP1 is a phosphoinositide effector and kinase scaffold. Adv. Biol. Regul. 2016, 60, 29–35.
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The biology of IQGAP proteins: Beyond the cytoskeleton. EMBO Rep. 2015, 16, 427–446.
- Sabo, Y.; de Los Santos, K.; Goff, S.P. IQGAP1 Negatively Regulates HIV-1 Gag Trafficking and Virion Production. Cell Rep. 2020, 30, 4065–4081.e4.
- Dolnik, O.; Kolesnikova, L.; Welsch, S.; Strecker, T.; Schudt, G.; Becker, S. Interaction with Tsg101 is necessary for the efficient transport and release of nucleocapsids in marburg virus-infected cells. PLoS Pathog. 2014, 10, e1004463.
- Gladue, D.P.; Holinka, L.G.; Fernandez-Sainz, I.J.; Prarat, M.V.; O’Donnell, V.; Vepkhvadze, N.G.; Lu, Z.; Risatti, G.R.; Borca, M.V. Interaction between Core protein of classical swine fever virus with cellular IQGAP1 protein appears essential for virulence in swine. Virology 2011, 412, 68–74.
- Lu, J.; Qu, Y.; Liu, Y.; Jambusaria, R.; Han, Z.; Ruthel, G.; Freedman, B.D.; Harty, R.N. Host IQGAP1 and Ebola virus VP40 interactions facilitate virus-like particle egress. J. Virol. 2013, 87, 7777–7780.
- Chen, D.; Wang, M.; Zhou, S.; Zhou, Q. HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J. 2002, 21, 6801–6810.
- Dumas, A.; Le-Bury, G.; Marie-Anais, F.; Herit, F.; Mazzolini, J.; Guilbert, T.; Bourdoncle, P.; Russell, D.G.; Benichou, S.; Zahraoui, A.; et al. The HIV-1 protein Vpr impairs phagosome maturation by controlling microtubule-dependent trafficking. J. Cell Biol. 2015, 211, 359–372.
- Stewart, S.A.; Poon, B.; Song, J.Y.; Chen, I.S. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J. Virol. 2000, 74, 3105–3111.
- Muthumani, K.; Hwang, D.S.; Desai, B.M.; Zhang, D.; Dayes, N.; Green, D.R.; Weiner, D.B. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J. Biol. Chem. 2002, 277, 37820–37831.
- Moon, H.S.; Yang, J.S. Role of HIV Vpr as a regulator of apoptosis and an effector on bystander cells. Mol. Cells 2006, 21, 7–20.
- Creagh, E.M.; Conroy, H.; Martin, S.J. Caspase-activation pathways in apoptosis and immunity. Immunol. Rev. 2003, 193, 10–21.
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727.
- Green, D.R. Caspase Activation and Inhibition. Cold Spring Harb. Perspect Biol. 2022, 14, a041020.
- Davis, M.J.; Swanson, J.A. Technical advance: Caspase-1 activation and IL-1beta release correlate with the degree of lysosome damage, as illustrated by a novel imaging method to quantify phagolysosome damage. J. Leukoc. Biol. 2010, 88, 813–822.
- Danielsson, F.; Peterson, M.K.; Caldeira Araujo, H.; Lautenschlager, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147.
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289.
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297.
- Hirata, Y.; Hosaka, T.; Iwata, T.; Le, C.T.; Jambaldorj, B.; Teshigawara, K.; Harada, N.; Sakaue, H.; Sakai, T.; Yoshimoto, K.; et al. Vimentin binds IRAP and is involved in GLUT4 vesicle trafficking. Biochem. Biophys. Res. Commun. 2011, 405, 96–101.
- Margiotta, A.; Bucci, C. Role of Intermediate Filaments in Vesicular Traffic. Cells 2016, 5, 20.
- Styers, M.L.; Salazar, G.; Love, R.; Peden, A.A.; Kowalczyk, A.P.; Faundez, V. The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol. Biol. Cell 2004, 15, 5369–5382.
- Ikawa, K.; Satou, A.; Fukuhara, M.; Matsumura, S.; Sugiyama, N.; Goto, H.; Fukuda, M.; Inagaki, M.; Ishihama, Y.; Toyoshima, F. Inhibition of endocytic vesicle fusion by Plk1-mediated phosphorylation of vimentin during mitosis. Cell Cycle 2014, 13, 126–137.
- Potokar, M.; Kreft, M.; Li, L.; Daniel Andersson, J.; Pangrsic, T.; Chowdhury, H.H.; Pekny, M.; Zorec, R. Cytoskeleton and vesicle mobility in astrocytes. Traffic 2007, 8, 12–20.
- Ivaska, J.; Vuoriluoto, K.; Huovinen, T.; Izawa, I.; Inagaki, M.; Parker, P.J. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005, 24, 3834–3845.
- Vardjan, N.; Gabrijel, M.; Potokar, M.; Svajger, U.; Kreft, M.; Jeras, M.; de Pablo, Y.; Faiz, M.; Pekny, M.; Zorec, R. IFN-gamma-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J. Neuroinflammation 2012, 9, 144.
- Potokar, M.; Stenovec, M.; Gabrijel, M.; Li, L.; Kreft, M.; Grilc, S.; Pekny, M.; Zorec, R. Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 2010, 58, 1208–1219.
- Zhang, Y.; Wen, Z.; Shi, X.; Liu, Y.J.; Eriksson, J.E.; Jiu, Y. The diverse roles and dynamic rearrangement of vimentin during viral infection. J. Cell Sci. 2020, 134, jcs250597.
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Perez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675.
- Helfand, B.T.; Mikami, A.; Vallee, R.B.; Goldman, R.D. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J. Cell Biol. 2002, 157, 795–806.
- Gyoeva, F.K.; Gelfand, V.I. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature 1991, 353, 445–448.
- Yoon, M.; Moir, R.D.; Prahlad, V.; Goldman, R.D. Motile properties of vimentin intermediate filament networks in living cells. J. Cell Biol. 1998, 143, 147–157.
- Prahlad, V.; Yoon, M.; Moir, R.D.; Vale, R.D.; Goldman, R.D. Rapid movements of vimentin on microtubule tracks: Kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 1998, 143, 159–170.
- Liao, G.; Gundersen, G.G. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J. Biol. Chem. 1998, 273, 9797–9803.
- Robert, A.; Herrmann, H.; Davidson, M.W.; Gelfand, V.I. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases. FASEB J. 2014, 28, 2879–2890.
- Hookway, C.; Ding, L.; Davidson, M.W.; Rappoport, J.Z.; Danuser, G.; Gelfand, V.I. Microtubule-dependent transport and dynamics of vimentin intermediate filaments. Mol. Biol. Cell 2015, 26, 1675–1686.
- Clarke, E.J.; Allan, V. Intermediate filaments: Vimentin moves in. Curr. Biol. 2002, 12, R596–R598.
- Aloisi, A.L.; Bucci, C. Rab GTPases-cargo direct interactions: Fine modulators of intracellular trafficking. Histol. Histopathol. 2013, 28, 839–849.
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34.
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small GTPase Rab7a. Biochim. Biophys. Acta 2013, 1833, 1283–1293.
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through Rac1 and vimentin. Biochim. Biophys. Acta Mol. Cell Res 2017, 1864, 367–381.
- Romano, R.; Del Fiore, V.S.; Bucci, C. Role of the Intermediate Filament Protein Peripherin in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15416.
- Fay, N.; Pante, N. The intermediate filament network protein, vimentin, is required for parvoviral infection. Virology 2013, 444, 181–190.
- Risco, C.; Rodriguez, J.R.; Lopez-Iglesias, C.; Carrascosa, J.L.; Esteban, M.; Rodriguez, D. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 2002, 76, 1839–1855.
- Sabharwal, P.; Amritha, C.K.; Sushmitha, C.; Natraj, U.; Savithri, H.S. Intracellular trafficking and endocytic uptake pathway of Pepper vein banding virus-like particles in epithelial cells. Nanomedicine 2019, 14, 1247–1265.
- Wu, W.; Pante, N. Vimentin plays a role in the release of the influenza A viral genome from endosomes. Virology 2016, 497, 41–52.
- Fernandez-Ortega, C.; Ramirez, A.; Casillas, D.; Paneque, T.; Ubieta, R.; Dubed, M.; Navea, L.; Castellanos-Serra, L.; Duarte, C.; Falcon, V.; et al. Identification of Vimentin as a Potential Therapeutic Target against HIV Infection. Viruses 2016, 8, 98.
- Koths, K.; Taylor, E.; Halenbeck, R.; Casipit, C.; Wang, A. Cloning and characterization of a human Mac-2-binding protein, a new member of the superfamily defined by the macrophage scavenger receptor cysteine-rich domain. J. Biol. Chem. 1993, 268, 14245–14249.
- Groschel, B.; Braner, J.J.; Funk, M.; Linde, R.; Doerr, H.W.; Cinatl, J., Jr.; Iacobelli, S. Elevated plasma levels of 90K (Mac-2 BP) immunostimulatory glycoprotein in HIV-1-infected children. J. Clin. Immunol. 2000, 20, 117–122.
- Tinari, N.; Natoli, C.; D’Ostilio, N.; Ghinelli, F.; Sighinolfi, L.; Ortona, L.; Tamburrini, E.; Piazza, M.; Chirianni, A.; Guerra, L.; et al. 90K (Mac-2 BP) predicts CD4 decline in human immunodeficiency virus-infected patients with CD4 counts above 200 × 10(6) cells/L. Arch. Pathol. Lab. Med. 1998, 122, 178–181.
- Darcissac, E.C.; Vidal, V.; De La Tribonniere, X.; Mouton, Y.; Bahr, G.M. Variations in serum IL-7 and 90K/Mac-2 binding protein (Mac-2 BP) levels analysed in cohorts of HIV-1 patients and correlated with clinical changes following antiretroviral therapy. Clin. Exp. Immunol. 2001, 126, 287–294.
- Wang, Q.; Zhang, X.; Han, Y.; Wang, X.; Gao, G. M2BP inhibits HIV-1 virion production in a vimentin filaments-dependent manner. Sci. Rep. 2016, 6, 32736.
- Adamson, C.S.; Freed, E.O. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv. Pharmacol. 2007, 55, 347–387.
- Jia, X.; Liu, L.; Liu, X.; Wu, D.; Yin, L.; Liu, X.; Zhang, J.; Yang, P.; Lu, H.; Zhang, L. Vimentin-a potential biomarker for therapeutic efficiency of HAART. Acta Biochim. Biophys. Sin. 2014, 46, 1001–1006.
- Shoeman, R.L.; Honer, B.; Stoller, T.J.; Kesselmeier, C.; Miedel, M.C.; Traub, P.; Graves, M.C. Human immunodeficiency virus type 1 protease cleaves the intermediate filament proteins vimentin, desmin, and glial fibrillary acidic protein. Proc. Natl. Acad. Sci. USA 1990, 87, 6336–6340.
- Shoeman, R.L.; Mothes, E.; Honer, B.; Kesselmeier, C.; Traub, P. Effect of human immunodeficiency virus type 1 protease on the intermediate filament subunit protein vimentin: Cleavage, in vitro assembly and altered distribution of filaments in vivo following microinjection of protease. Acta Histochem. Suppl. 1991, 41, 129–141.
- Shoeman, R.L.; Mothes, E.; Kesselmeier, C.; Traub, P. Intermediate filament assembly and stability in vitro: Effect and implications of the removal of head and tail domains of vimentin by the human immunodeficiency virus type 1 protease. Cell Biol. Int. Rep. 1990, 14, 583–594.
- Karczewski, M.K.; Strebel, K. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 1996, 70, 494–507.
- Shoeman, R.L.; Honer, B.; Mothes, E.; Traub, P. Potential role of the viral protease in human immunodeficiency virus type 1 associated pathogenesis. Med. Hypotheses 1992, 37, 137–150.
- Armenian, H.K.; Hoover, D.R.; Rubb, S.; Metz, S.; Martinez-Maza, O.; Chmiel, J.; Kingsley, L.; Saah, A. Risk factors for non-Hodgkin’s lymphomas in acquired immunodeficiency syndrome (AIDS). Am. J. Epidemiol 1996, 143, 374–379.
- Chou, K.C.; Tomasselli, A.G.; Reardon, I.M.; Heinrikson, R.L. Predicting human immunodeficiency virus protease cleavage sites in proteins by a discriminant function method. Proteins 1996, 24, 51–72.