The number of patients with nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) is increasing globally and is raising serious concerns regarding the increasing medical and economic burden incurred for their treatment. The progression of NASH to more severe conditions such as cirrhosis and hepatocellular carcinoma requires liver transplantation to avoid death. Therefore, therapeutic intervention is required in the NASH stage, although no therapeutic drugs are currently available for this. Several anti-NASH candidate drugs have been developed that enable treatment via the modulation of distinct signaling cascades and include a series of drugs targeting peroxisome proliferator-activated receptor (PPAR) subtypes (PPARα/δ/γ) that are considered to be attractive because they can regulate both systemic lipid metabolism and inflammation.
- lanifibranor
- saroglitazar
- bezafibrate
- pemafibrate
- PPAR
- dual/pan agonist
- X-ray crystallography
1. Lanifibranor (Peroxisome Proliferator-Activated Receptor Pan Agonist)—Under Consideration for Treating Nonalcoholic Fatty Liver Disease/Non-Alcoholic Steatohepatitis
2. Chiglitazar (Peroxisome Proliferator-Activated Receptor Pan Agonist)—Under Consideration in China
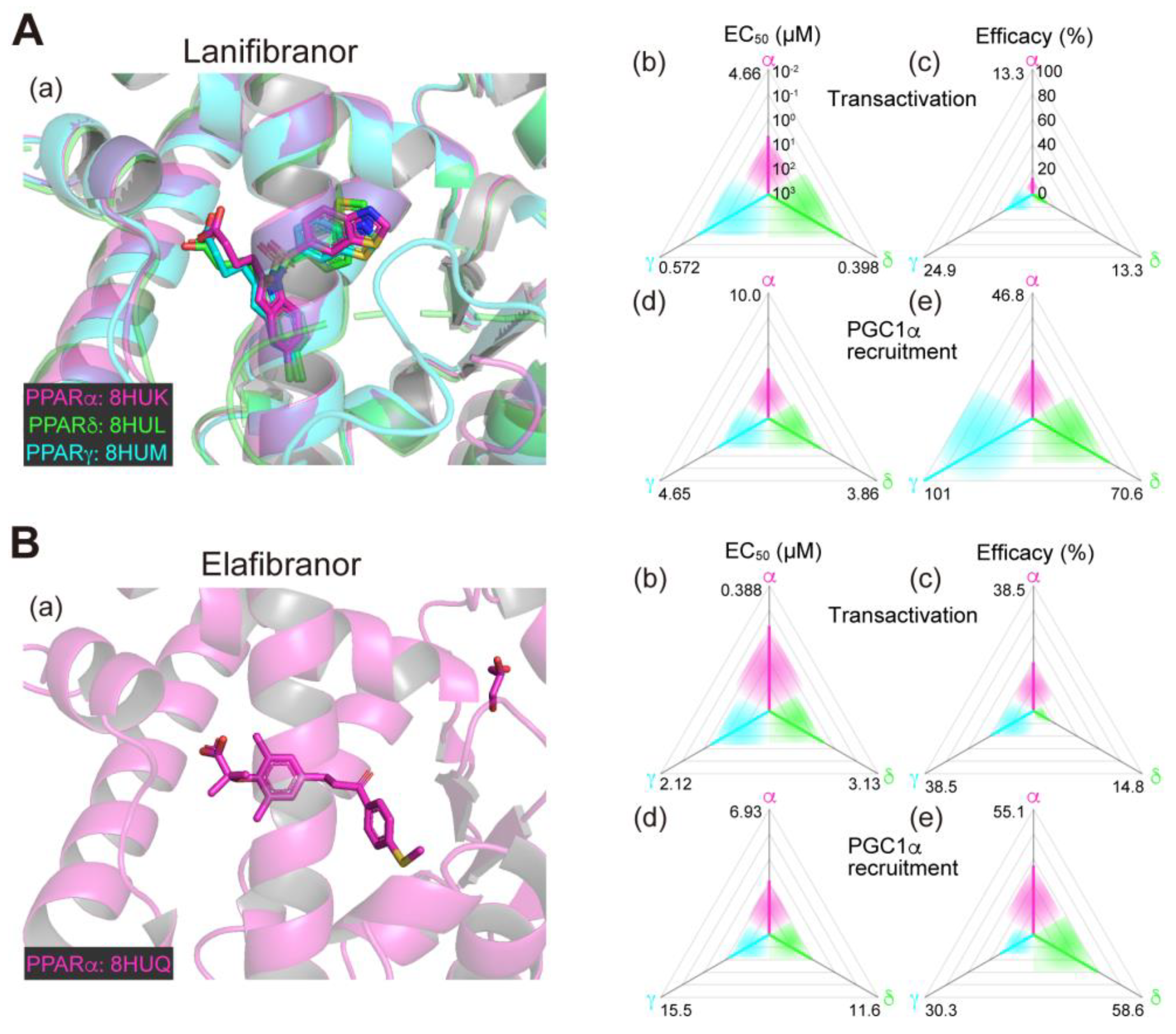
3. Elafibranor (PPARα/δ Dual Agonist)—Discontinued
4. Saroglitazar (PPARα/γ Dual Agonist)—Under Consideration
5. Seladelpar (PPARδ-Selective Agonist)—Interrupted
6. Fenofibrate (PPARα(/γ Dual) Agonist)—Discontinued
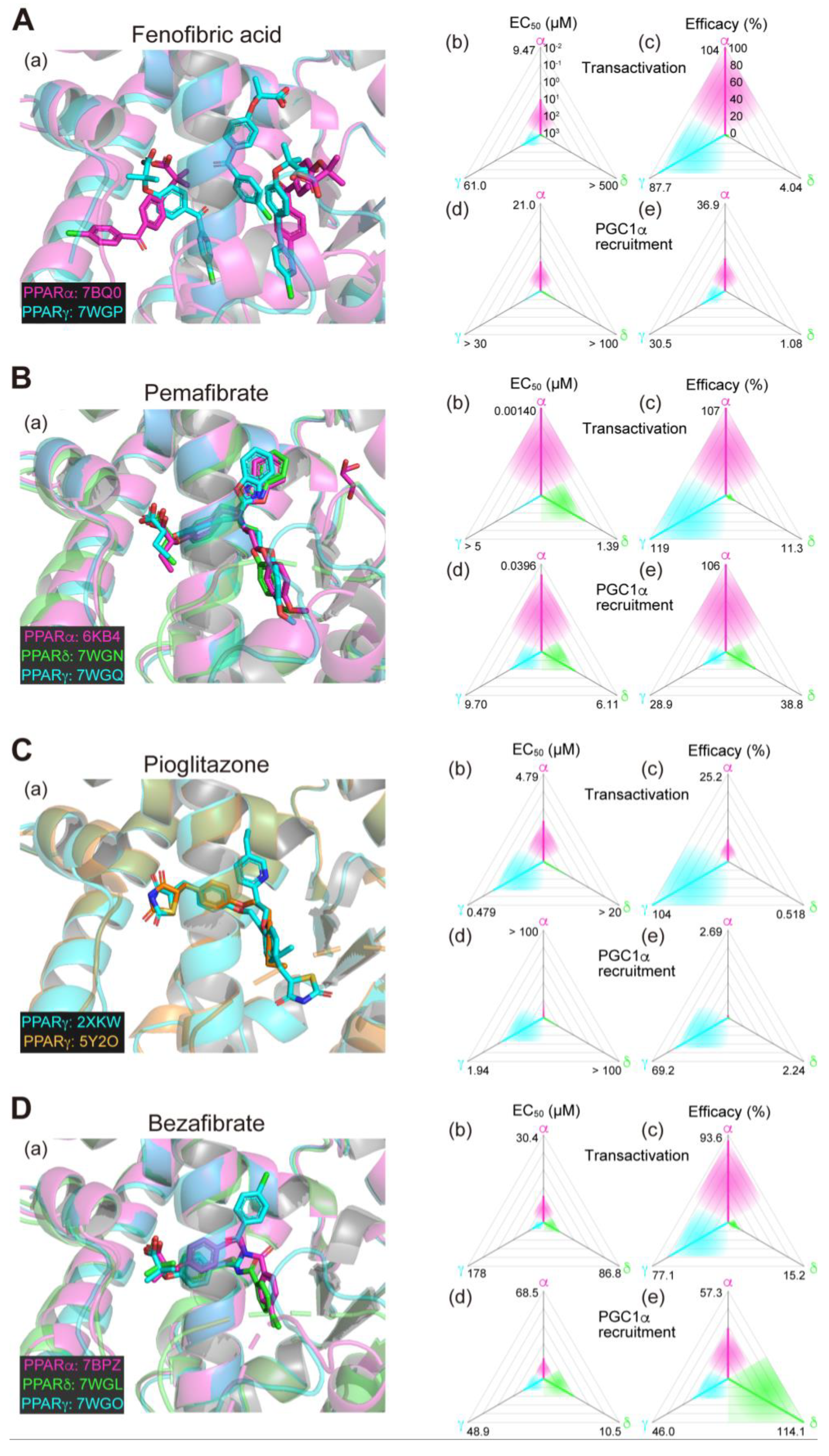
7. Pemafibrate (PPARα-Selective Agonist)—Under Consideration in Japan
8. Pioglitazone (PPARγ-Selective Agonist)—Under Consideration
9. Rosiglitazone (PPARγ-Selective Agonist)—Discontinued
10. Lobeglitazone (PPARγ Agonist)—Under Consideration in Korea
This entry is adapted from the peer-reviewed paper 10.3390/biom13081264
References
- Wettstein, G.; Luccarini, J.M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrêne, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537.
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) α/γ/δ triple activators: Discovery of lanifibranor, a new antifibrotic clinical candidate. J. Med. Chem. 2018, 61, 2246–2265.
- Honda, A.; Kamata, S.; Akahane, M.; Machida, Y.; Uchii, K.; Shiiyama, Y.; Habu, Y.; Miyawaki, S.; Kaneko, C.; Oyama, T.; et al. Functional and structural insights into human PPARα/δ/γ subtype selectivity of bezafibrate, fenofibric acid, and pemafibrate. Int. J. Mol. Sci. 2022, 23, 4726.
- Honda, A.; Kamata, S.; Satta, C.; Machida, Y.; Uchii, K.; Terasawa, K.; Nemoto, A.; Oyama, T.; Ishii, I. Structural basis for anti-non-alcoholic fatty liver disease and diabetic dyslipidemia drug saroglitazar as a PPAR α/γ dual agonist. Biol. Pharm. Bull. 2021, 44, 1210–1219.
- Kamata, S.; Honda, A.; Ishii, I. Functional and structural insights into the human PPARα/δ/γ targeting preferences of anti-NASH investigational drugs, lanifibranor, seladelpar, and elafibranor. Antioxidants 2023, 12, 1523.
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727.
- Sven, M.F.; Pierre, B.; Manal, F.A.; Quentin, M.A.; Elisabetta, B.; Vlad, R.; Philippe, H.M.; Bruno, S.; Jean-Louis, J.; Pierre, B.; et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: Design of the NATIVE study. Contemp. Clin. Trials 2020, 98, 106170.
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558.
- Deeks, E.D. Chiglitazar: First approval. Drugs 2022, 82, 87–92.
- Kamata, S.; Oyama, T.; Ishii, I. Preparation of co-crystals of human PPARα-LBD and ligand for high-resolution X-ray crystallography. STAR Protoc. 2021, 2, 100364.
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952.
- Tølbøl, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194.
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159.e5.
- GENFIT Press Release (11 May 2020). GENFIT: Announces Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis. Available online: https://ir.genfit.com/news-releases/news-release-details/genfit-announces-results-interim-analysis-resolve-it-phase-3/ (accessed on 25 July 2023).
- Jani, R.H.; Kansagra, K.; Jain, M.R.; Patel, H. Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects. Clin. Drug Investig. 2013, 33, 809–816.
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist–Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80.
- Mitra, A. An observational study of reduction in glycemic parameters and liver stiffness by saroglitazar 4 mg in patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Cureus 2020, 12, e9065.
- Roy, S. Clinical case series of decrease in shear wave elastography values in ten diabetic dyslipidemia patients having NAFLD with saroglitazar 4 mg: An Indian experience. Case Rep. Med. 2020, 2020, 4287075.
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: A randomized controlled double-blind phase 2 trial. Hepatology 2021, 74, 1809–1824.
- CymaBay Therapeutics Press Release (25 November 2019). CymaBay Therapeutics Halts Clinical Development of Seladelpar. Available online: https://ir.cymabay.com/press-releases/detail/476/cymabay-therapeutics-halts-clinical-development-of-seladelpar (accessed on 25 July 2023).
- CymaBay Therapeutics Press Release (23 July 2020). FDA Lifts All Clinical Holds on Seladelpar. Available online: https://ir.cymabay.com/press-releases/detail/485/fda-lifts-all-clinical-holds-on-seladelpar (accessed on 25 July 2023).
- Oscarsson, J.; Önnerhag, K.; Risérus, U.; Sundén, M.; Johansson, L.; Jansson, P.A.; Moris, L.; Nilsson, P.M.; Eriksson, J.W.; Lind, L. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 2018, 12, 1390–1403.e4.
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a new selective PPARα modulator: Drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr. Atheroscler. Rep. 2020, 22, 5.
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477.
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.C.; et al. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818.
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 2021, 54, 1263–1277.
- Fruchart, J.C.; Hermans, M.P.; Fruchart-Najib, J.; Kodama, T. Selective peroxisome proliferator-activated receptor alpha modulators (SPPARMα) in the metabolic syndrome: Is pemafibrate light at the end of the tunnel? Curr. Atheroscler. Rep. 2021, 23, 3.
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152.
- Kusunoki, M.; Sakazaki, T.; Tsutsumi, K.; Miyata, T.; Oshida, Y. The effects of pemafibrate in Japanese patients with type 2 diabetes receiving HMG-CoA reductase inhibitors. Endocr. Metab. Immune. Disord. Drug Targets 2021, 21, 919–924.
- Lebovitz, H.E. Thiazolidinediones: The forgotten diabetes medications. Curr. Diab. Rep. 2019, 19, 151.
- Lee, M.A.; Tan, L.; Yang, H.; Im, Y.G.; Im, Y.J. Structures of PPARγ complexed with lobeglitazone and pioglitazone reveal key determinants for the recognition of antidiabetic drugs. Sci. Rep. 2017, 7, 16837.
- Tokushige, K.; Ikejima, K.; Ono, M.; Eguchi, Y.; Kamada, Y.; Itoh, Y.; Akuta, N.; Yoneda, M.; Iwasa, M.; Yoneda, M.; et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis 2020. J. Gastroenterol. 2021, 56, 951–963.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685.
- Huang, J.F.; Dai, C.Y.; Huang, C.F.; Tsai, P.C.; Yeh, M.L.; Hsu, P.Y.; Huang, S.F.; Bair, M.J.; Hou, N.J.; Huang, C.I.; et al. First-in-Asian double-blind randomized trial to assess the efficacy and safety of insulin sensitizer in nonalcoholic steatohepatitis patients. Hepatol. Int. 2021, 15, 1136–1147.
- Jacques, V.; Bolze, S.; Hallakou-Bozec, S.; Czarnik, A.W.; Divakaruni, A.S.; Fouqueray, P.; Murphy, A.N.; Van der Ploeg, L.H.T.; DeWitt, S. Deuterium-stabilized (R)-pioglitazone (PXL065) is responsible for pioglitazone efficacy in NASH yet exhibits little to no PPARγ activity. Hepatol. Commun. 2021, 5, 1412–1425.
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471.
- Singh, S.; Loke, Y.K.; Furberg, C.D. Long-term risk of cardiovascular events with rosiglitazone: A meta-analysis. JAMA 2007, 298, 1189–1195.
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110.
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453.
- Bae, J.; Park, T.; Kim, H.; Lee, M.; Cha, B.S. Lobeglitazone: A novel thiazolidinedione for the management of type 2 diabetes mellitus. Diabetes Metab. J. 2021, 45, 326–336.
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: Its efficacy and predictive factors related to responsiveness. J. Korean Med. Sci. 2017, 32, 60–69.