Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Neuroglobin, which is a heme protein from the globin family that is predominantly expressed in nervous tissue, can promote a neuronal survivor. However, the molecular mechanisms underlying the neuroprotective function of Ngb remain poorly understood to this day. The interactions between neuroglobin and mitochondrial cytochrome c may serve as at least one of the mechanisms of neuroglobin-mediated neuroprotection.
- neuroglobin
- neuroprotection
- cytochrome c
- heme proteins
1. Introduction
Neuroglobin (Ngb) is a six-coordinate heme protein from the globin family that is predominantly expressed in nervous tissue [1]. Intracellular Ngb concentrations range from 100 μM in the neurons of the hypothalamus and the retinal cells to 1 μM in the other neurons [1,2,3]. Furthermore, there is an average 2-5-fold increase in NGB gene expression in response to various stress signals (e.g., hypoxia or oxidative stress) [4,5]. Ngb is a cytoplasmic protein, although there is evidence for Ngb localization not only in close proximity to mitochondria, but also within mitochondria [6,7,8]. However, Ngb lacks a mitochondrial target sequence, and the exact mechanism regulating Ngb mitochondrial localization is still unclear [9].
Ngb is a monomer with a molecular weight of about 17 kDa, consists of eight α-helices (A-H), and has a typical three-over-three globin fold [1,10,11]. Ngb contains one heme group, and, in the absence of exogenous ligands, the axial coordination positions of the heme are occupied by two histidine residues (His64-Fe-His96) [11,12]. Human Ngb features three cysteine residues (Cys46, Cys55, and Cys120), two of which form an intramolecular disulfide bridge (Cys46–Cys55) [1,10,11]. The formation of other intramolecular disulfide bridges in Ngb involving Cys120 and Cys46 or Cys55 is considered to be sterically impossible [11,13]. The significance of the Cys46–Cys55 disulfide bridge is determined by its effect on the conformation of the CD-loop (the region between α-helices C and D) and modulation of Ngb functionality. Upon cleavage of this disulfide bridge, the bond between the heme iron and His64 is strengthened, thus resulting in a ten-fold decrease of the His64 dissociation rate. As a consequence, the affinity of Ngb for exogenous ligands is reduced because the dissociation of His64 from the heme is a necessary and rate-determining step for the binding of exogenous ligands to Ngb [11,14]. However, it is suggested that, in vivo, more than 88% of Ngb molecules are in the ferrous deoxy form without a disulfide bridge [15,16,17].
Similar to hemoglobin and myoglobin, Ngb reversibly binds O2 and other small gaseous ligands [12,18]. Ngb catalyzes the NO deoxygenation reaction, thus yielding NO3− [19,20], as well as NO generation from NO2− (nitrite reductase activity) [21,22,23].
Ngb has been shown to promote neuronal survival in conditions such as ischemia, hypoxia, Alzheimer’s and Huntington’s diseases, oxidative stress, stroke, spinal cord injury, retinal degeneration, arsenic poisoning, etc., in numerous in vitro and in vivo studies [4,5,9,24,25,26,27,28,29]. However, the molecular mechanisms underlying the neuroprotective function of Ngb remain poorly understood to this day. Because Ngb can bind O2, it was initially assumed that Ngb, like myoglobin, acts as an O2 depot and transporter under hypoxia [1,2,3]. However, relatively low cellular concentrations of Ngb and a very rapid autoxidation contradict this assumption [30].
Recent hypotheses suggest that Ngb-mediated neuroprotection is based on its involvement in various biochemical cascades of the cell [4,9,24,28,29]. Ngb can detoxify reactive oxygen and nitrogen species [31,32,33,34], and there is evidence of Ngb involvement in the Wnt/β-catenin pathway [35,36], as well as in the PI3K/Akt/MAPK signaling pathway [37,38]. Additionally, the protein–protein interactions between Ngb and voltage-dependent anion channels (VDACs) [6,7,39], α-subunits of heterotrimeric G-protein [40,41,42], and mitochondrial cytochrome c (Cyt c) [30,43] may also contribute to Ngb-mediated neuroprotection. This review is focused on the molecular interactions between Ngb and Cyt c.
Mitochondrial Cyt c, a multifunctional heme protein, plays a crucial role as an electron transporter in the respiratory chain. During the activation of apoptosis, Cyt c translocates from the mitochondria into the cytoplasm, where it amplifies the external apoptotic signal or initiates the caspase cascade through the intrinsic pathway (making it Cyt c-dependent) [44,45,46,47]. In addition to physiological role of apoptosis in nervous tissue, it is also associated with pathological conditions, such as neurodegenerative diseases, strokes, ischemia, hypoxia, etc. Furthermore, the initiation of apoptosis occurs under the influence of various stress stimuli, including pathogens, radiation, chemotherapeutic drugs, oxidative stress, etc., which cause mitochondrial dysfunction [48,49,50].
The key event of the intrinsic apoptotic pathway is the outer mitochondrial membrane permeabilization, which leads to the release of various proapoptotic factors, including Cyt c, into the cytoplasm. Furthermore, the activation of the caspase cascade depends on the efficiency of Cyt c binding to apoptotic protease activating factor-1 (Apaf-1), thereby leading to apoptosome assembly. It should be noted that only ferric Cyt c can bind to Apaf-1, thereby activating apoptosome assembly and triggering the caspase cascade [51].
The hypothesis about the interaction between Ngb and Cyt c was proposed based on the kinetic studies of the redox reaction between Ngb(Fe2+) and Cyt c(Fe3+):
This reaction was initially observed by absorbance spectroscopy under anaerobic conditions [43]. According to this hypothesis, Ngb(Fe2+) reacts with Cyt c(Fe3+) when the latter translocates into the cytoplasm during the activation of the intrinsic apoptotic pathway. This reaction prevents Cyt c(Fe3+) from interacting with Apaf-1 and, thus, prevents apoptosome assembly and ultimately blocks the initiation of apoptosis [30,43,52]. Later, it was hypothesized that Ngb can potentially interact with Cyt c(Fe3+), regardless of its redox state [53,54].
Ngb(Fe2+) + Cyt c(Fe3+) → Ngb(Fe3+) + Cyt c(Fe2+).
Therefore, Ngb can protect neurons from apoptosis by reducing the concentration of cytosolic Cyt c(Fe3+), which can increase under conditions that cause mitochondrial dysfunction and during normal mitochondrial function (reducing “accidental” Cyt c release) [30].
2. Electron Transfer between Ferrous Neuroglobin and Ferric Cytochrome c
The investigation of interactions between Ngb and Cyt c began with the observation of the very rapid electron transfer from recombinant murine Ngb(Fe2+) to Cyt c(Fe3+) [43]. The second-order rate constant for the reaction (k = 2 × 107 M−1 s−1) is close to the known physiologically significant Cyt c redox reactions rate constants, e.g., the reduction of cytochrome c oxidase or oxidation of cytochrome b5 [43,46,55]. It should be noted that, unlike human Ngb, murine Ngb lacks the disulfide bridge due to the replacement of Cys46 by Gly [1]. Hence, it can be concluded that the presence of the Ngb disulfide bridge does not affect electron transfer from Ngb(Fe2+) to Cyt c(Fe3+) [43].
However, a recent study [56] has come to somewhat controversial conclusions. The electron transfer between recombinant human Ngb(Fe2+) and Cyt c(Fe3+) was studied using a nanoporous gold electrode. The immobilization of Ngb onto the electrode surface enabled the rapid reduction of the Ngb heme, after which Cyt c(Fe3+) was added to the solution, and its reduction to Cyt c(Fe2+) was observed. In the case of the Ngb C55S mutant without the disulfide bridge (Cys46–Cys55), there were almost no voltammogram changes upon Cyt c(Fe3+) addition. Thus, it was concluded that the disulfide bridge has significance for electron transfer between Ngb(Fe2+) and Cyt c(Fe3+). These results are inconsistent with the previously obtained data on electron transfer between murine Ngb(Fe2+) and Cyt c(Fe3+) [43]. It is assumed that the Ngb disulfide bridge formation under oxidative stress conditions is potentially capable of modulating the functionality of Ngb. For instance, we can consider the electron transfer to Cyt c(Fe3+) in response to redox changes in the cell [14,18,56].
Electron transfer between Ngb(Fe2+) and Cyt c(Fe3+) was also detected by stopped-flow spectroscopy [57], while recombinant human Ngb was reduced with an excess of sodium dithionite, which most likely led to the disulfide bridge cleavage as well.
Based on the aforementioned data, two hypotheses regarding the interaction between Ngb(Fe2+) and Cyt c(Fe3+) in vivo were formed [30,43,56]. According to one of them [30,43,52], Ngb(Fe2+) in the ferrous deoxy form, which normally prevails in cells [15,16,17], reduces Cyt c(Fe3+) molecules by leaving the mitochondria into the cytoplasm to Cyt c(Fe2+), thereby preventing apoptosome assembly and apoptosis triggering along the Cyt c-dependent pathway (Figure 1, I). It should be noted that such a mechanism takes place under normal cellular conditions, thus preventing the consequences of the “accidental” Cyt c(Fe3+) release from mitochondria [30], which is a constitutive process associated with transient VDAC pore opening [58,59,60]. Even through “accidental” Cyt c(Fe3+) release is a small-scale process, Cyt c(Fe3+) can bind to the inositol-1,4,5-triphosphate receptors on the endoplasmic reticulum, thus causing Ca2+ release to the cytoplasm. An increase in the level of cytosolic Ca2+ further stimulates Cyt c(Fe3+) release from the mitochondria, thus establishing an ongoing amplification signal of the initial Cyt c(Fe3+) release [30,61]. Therefore, the existence of a resetting mechanism for the threshold level of Cyt c(Fe3+), for instance, via redox reaction with Ngb(Fe2+), is highly probable [30]. This hypothesis also explains the predominant localization of Ngb in neurons and retinal cells at high concentrations [2,3]. These highly specialized and metabolically active cells experience frequent high fluxes of cellular Ca2+ during their normal physiological functioning. As a result, these cell types might have a tendency to undergo programmed cell death as a result of the “accidental” release of Cyt c(Fe3+) in the cytoplasm. Hence, higher cellular concentrations of Ngb are required to protect these cells from apoptosis [30]. This hypothesis is further supported by the data on the Ngb localization in close proximity to mitochondria [7]. It is important to note that, in accordance with this mechanism, Ngb can suppress the triggering of the apoptotic process through the “accidental” release of Cyt c(Fe3+) while still allowing committed programmed cell death to occur under appropriate circumstances [30].
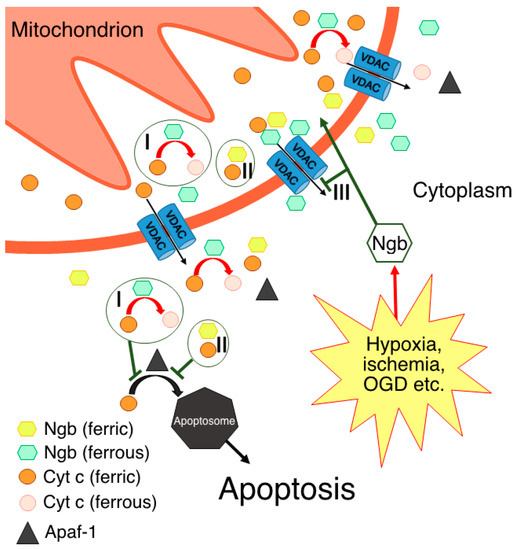
Figure 1. Possible molecular mechanisms of Ngb-mediated neuroprotection through direct or indirect interactions with Cyt c: I—electron transfer reaction, thus resulting in Cyt c being unable to form apoptosome via its interaction with Apaf-1; II—formation of complex between ferric forms of Ngb and Cyt c; III—prevention of Cyt c release in the cytoplasm via Ngb interaction with VDACs.
According to another hypothesis [56], electron transfer from Ngb(Fe2+) to Cyt c(Fe3+) occurs only in the presence of the disulfide bridge in the Ngb structure, which corresponds to the oxidative stress conditions [14,18,56]. In support of this idea, NGB gene expression is upregulated in response to various stress signals [4,5]. Moreover, although Ngb cellular concentration is primarily high only in neurons and retinal cells [2,3], it can increase in other cell types under cellular stress conditions to a level that is sufficient for Ngb-mediated protection against apoptosis [4,5]. This hypothesis contradicts the one described above, but not vice versa. It is quite probable that electron transfer from Ngb(Fe2+) to Cyt c(Fe3+) is possible in both cases: from Ngb(Fe2+) with the disulfide bridge and from Ngb(Fe2+) with reduced cysteines. In this case, the disulfide bridge may play a crucial role in modulating this electron transfer reaction under the corresponding redox conditions. It should be noted that, under oxidative stress conditions, not only cysteine residues, but also the heme of Ngb can be oxidized, which will lead to the inability of the Ngb to act as electron donor for the Cyt c. In addition, a Ngb-reducing system in vivo is still unknown, although several attempts have been made to identify it [43,62,63]. Hence, in the case of Ngb heme oxidation, the interaction with Cyt c is either disrupted or follows other mechanisms without electron transfer [54].
Thus, the studies on electron transfer between Ngb(Fe2+) and Cyt c(Fe3+) are rather inconsistent and limited. This inconsistency arises from the experimental difficulties, such as the preparation of pure ferrous Ngb without a reducing agent as a consequence of Ngb’s high autoxidation rate (0.23 ± 0.03 min−1 [17]). The reducing agent molecules can compete with Ngb(Fe2+) for the reduction of Cyt c(Fe3+) molecules in the solution; thus, the possibility of false positive results originates. Another challenge is the inability to use a classical strategy for the study of the electron transfer between cytochromes and heme globins. This strategy utilizes the difference in CO binding between six-coordinated cytochromes and five-coordinated heme globins, which cannot be used given the six-coordination nature of Ngb [57]. It is evident that further studies of the electron transfer between Ngb(Fe2+) and Cyt c(Fe3+) with alternative approaches are required.
This entry is adapted from the peer-reviewed paper 10.3390/biom13081233
This entry is offline, you can click here to edit this entry!