The genomes of polyomaviruses are characterized by their tripartite organization with an early region, a late region and a noncoding control region (NCCR). The early region encodes proteins involved in replication and transcription of the viral genome, while expression of the late region generates the capsid proteins. Transcription regulatory sequences for expression of the early and late genes, as well as the origin of replication are encompassed in the NCCR. Cell tropism of polyomaviruses not only depends on the appropriate receptors on the host cell, but cell-specific expression of the viral genes is also governed by the NCCR.
- disease
- Merkel cell carcinoma
- mutation
- NCCR
- novel human polyomaviruses
- transcription factor binding sites
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction: Human Polyomaviruses
Polyomaviruses (PyVs) are non-enveloped viruses that are typically 40–45 nm in diameter, and that possess a double-stranded circular genome of around 5000 base-pairs. Birds and mammals, including humans, are natural hosts for PyVs [1,2]. Recently, PyVs have also been isolated from fish [3,4]. So far, 15 different polyomaviruses have been isolated from human samples. The first human polyomaviruses, BKPyV and JCPyV, were identified in 1971 [5,6]. In 2007, two new human polyomaviruses (Karolinska Institute PyV (KIPyV) [7] and Washington University PyV (WUPyV) [8] were detected, and in the following years, Merkel cell PyV (MCPyV) [9], HPyV6 [10], HPyV7 [10], Trichodisplasia spinulosa PyV (TSPyV) [11], HPyV9 [12], HPyV10 [13], Saint Louis PyV (STLPyV) [14], HPyV12 [15], New Jersey PyV (NJPyV) [16], Lyon IARC PyV (LIPyV) [17], and Quebec PyV [18] have been described. Their original source of isolation and association with human diseases is summarized in Table 1.
Table 1. The novel human polyomaviruses, their original source of isolation and their association with human diseases.
|
Virus |
Original Source |
Associated Disease |
Reference |
|
KIPyV |
Nasopharyngeal aspirate |
None |
[7] |
|
WUPyV |
Bronchoavelar lavage |
None |
[8] |
|
MCPyV |
Merkel cell carcinoma |
None |
[9] |
|
HPyV6 |
Healthy skin |
Pruritic skin eruption in immunocompromised patients |
[10] |
|
HPyV7 |
Healthy skin |
Pruritic skin eruption in immunocompromised patients |
[10] |
|
TSPyV |
Trichodysplasia spinulosa spicules |
Trichodysplasia spinulosa |
[11] |
|
HPyV9 |
Serum from renal transplant recipient |
None |
[12] |
|
HPyV10 |
Condyloma specimens from a patient with WHIM * syndrome |
None |
[13] |
|
STLPyV |
Stool sample from a healthy 15-month-old child |
None |
[14] |
|
HPyV12 |
Liver sample from patient with malignant disease |
None |
[15] |
|
NJPyV |
Muscle biopsy from a pancreatic transplant patient |
None |
[16] |
|
LIPyV |
Skin swab |
None |
[17] |
|
QPyV |
Stool sample from 85-year old hospital patient |
None |
[18] |
* warts, hypogammaglobulinemia, infections, and myelokathexis.
Whether all of these are genuine human polyomaviruses (HPyVs) remains to be determined. BKPyV, JCPyV, KIPyV, WUPyV, MCPyV, HPyV6, HPyV, TSPyV, HPyV9, HPyV10, STLPyV, HPyV12, and NJPyV are classified as human polyomaviruses by the International Committee of Taxonomy of Viruses [19,20], LIPyV has only been very recently described, while LIPyV DNA was originally detected in human skin [17]. LIPyV seroreactivity in healthy individuals is ~5% in healthy individuals and much lower than the seroprevalence of the other HPyVs, which is between 50–100% [21]. Accordingly, LIPyV DNA was not detected or present in <2% of examined skin, eyebrow hair, gargle samples and tonsil samples [17,22,23]. Moreover, LIPyV DNA was frequently detected in the feces of cats [24], suggesting that it may be a feline PyV rather than a HPyV. QPyV DNA was detected in the feces of one patient [18], and the seroprevalence of this PyV has not been examined. Despite its original identification in human liver, gastro-intestinal tract and colon tissue and a VP1 seropositivity (respectively LT seropositivity) between ~20–90% (respectively 30–40%) in healthy adults or malignant and non-malignant gastro-intestinal tract patients [15,25], HPyV12 DNA could not be detected in numerous human samples from different sources [22,26–30]. Moreover, the group of Feltkamp reported that HPyV12 seroprevalence is only around 5% [21,31]. A nearly identical HPyV12 variant was isolated from shrew, suggesting that HPyV12 may be transmitted from shrew to humans, or that human HPyV12 positive samples were contaminated [32].
2. The Polyomavirus Genome: The Noncoding Control Region
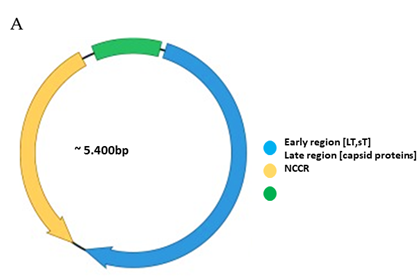
Functionally, the PyV genome is tripartite consisting of the early region, the late region, and the noncoding control region (NCCR) (Figure 1A). The early region codes for regulatory proteins involved in replication and transcription of the viral genome. The major early proteins are large T-antigen (LT) and small t-antigen (sT). The late region codes for the structural proteins VP1, VP2 and VP3 that form the capsid. VP1 is the major capsid protein, while VP2 and VP3 are the minor capsid proteins [1,2]. However, not all PyVs express VP3 [33]. Interspersed between the early and late region are sequences that do not code for viral proteins, and is referred to as the NCCR.
B
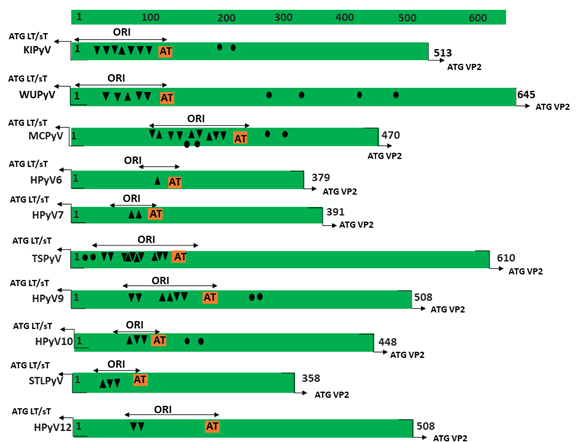
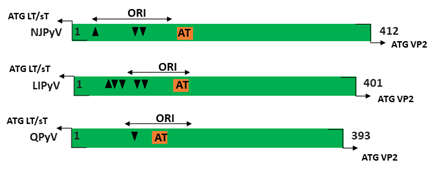
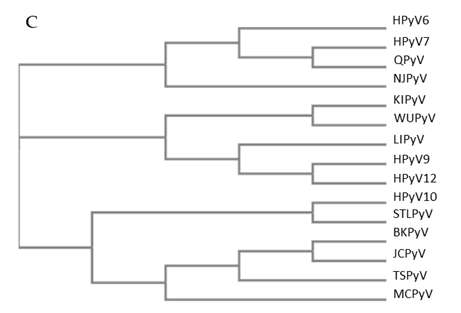
Figure 1. Genomic organization of the human polyomaviruses (HPyVs) genome and the structure of the noncoding control region (NCCR). (A) The circular dsDNA genome consists of the early and late regions that encode regulatory and structural proteins, respectively. Interspersed is the NCCR. (B) Schematic presentation of the NCCR of the novel HPyVs. The NCCR is the region between the start codon of Large T antigen (LT) and Small T antigen (sT) and the start codon of VP2. The AT-rich region (AT), repeated sequences (black dots), and LT binding motifs (upward pointing triangle = 5′-GRGGC-3′; downward pointing triangle = 5′-GCCYC-3′) are shown. (C) Phylogenetic tree bases on NCCR sequences of the different HPyVs. This is a neighbor-joining tree without distance corrections using Clustal Omega multiple sequence alignment [34].
Studies with simian virus 40 (SV40 or Macaca mulatta polyomavirus 1) and murine polyomaviruses have been pivotal in unveiling the functions of this region. The SV40 NCCR contains the origin of replication (ori), which consists of GRGGC motifs to which LT binds and is flanked by an AT-rich sequence and an easily denaturated imperfect palindrome [35,36]. Binding of LT to these motifs is also involved in regulation of viral transcription [37,38]. The NCCR also contains promoter and enhancer elements that control early and late transcription [39,40]. SV40 directly isolated from its natural host, rhesus monkey, has a NCCR that consists of an AT-rich region, triple GC-rich 21 base-pairs (bp) repeats, and a single 72 bp element. The 21 bp repeats contain the LT binding motif (GRGGC; [41,42]). This NCCR organization is known as the archetype. SV40 adapted to grow in cell culture has a duplication of this 72 bp element, with this type of NCCR referred to as prototype [43,44]. SV40 isolated from human tumors usually contain a single 72 bp repeat [43]. Rearrangements in the SV40 NCCR affect viral transcription and replication, as well as oncogenic properties of the virus [45,46]. The Mouse polyomavirus (Mus musculus polyomavirus 1; MPyV) NCCR encompasses the ori consisting of an AT-tract and a GC-rich (LT binding motifs) inverted repeat, and the transcription regulatory domains A (or α) and B (or β), C and D [47–50]. Alterations in the MPyV NCCR have an effect on viral replication in cell culture and in the host, the host range, and in vitro transformation [51–55].
The NCCR of the HPyVs varies between 267 bp (JCPyV CY-strain; accession number AB038249) to 645 bp (WUPyV prototype; accession number NC_009539) (see Figure S1 for the NCCR sequences of the novel HPyV), and similar to the NCCR of SV40 and MPyV, the NCCR of HPyVs also contain the origin of replication, LT binding motifs, and AT-rich region (Figure 1B). This region of the genome displays little or no sequence identity between the different HPyV species (Figure S2). A neighbor-joining tree without distance corrections shows which NCCRs are most closely related (Figure 1C).
The diversification based on the presence of a certain NCCR rearranged structure contributed to determining HPyVs strains as “archetype” or prototype”. The importance of the NCCR rearrangements during HPyVs infection became obvious when different strains of JCPyV were examined. The archetype JCPyV NCCR strain (CY) is divided into six boxes named A (36 bp), B (23 bp), C (55 bp), D (66 bp), E (18 bp), and F (69 bp) and contains the origin of replication (ORI), the promoter and the enhancer elements [56]. The NCCR harbored transcription factor binding sites such as the nuclear transcription factor-1 (NF1), a JCPyV cell-specific regulator of promoter and enhancer activity [57,58], the activating protein 1 (AP1), involved in JCPyV early gene expression [57,59], and the specificity protein-1 (SP1) able to regulate JCPyV transcription [57,60]. The archetype NCCR is considered the transmissible form of the virus among the population, and could be released into the urine of healthy individuals due to periodic and subclinical reactivation in the kidney [61,62]. In contrast, in the context of immunosuppression or during immunomodulatory therapy or in AIDS patients, JCPyV can reactivate from latency to cause a fatal pathology of the central nervous system (CNS), known as progressive multifocal leukoencephalopathy (PML) [61]. JCPyV variants carrying rearranged NCCR were usually isolated from PML patients. The prototype Mad-1 strain is the most studied variant of JCPyV and is characterized by 98-bp tandem repeats in the NCCR late proximal region (arranged as ORI-A-C-E-A-C-E-F), and is able to increase viral gene expression in human glial cells, thereby indicating that it is involved in controlling cell gene expression [63–65]. The enhancer repeats found in the Mad-1 strain are lacking in the archetype JCPyV strains isolated from the urine of healthy individuals [64]. Additional NCCR rearrangements are implicated in the development of the JCPyV pathogenic strains. In fact, in a significant proportion of JCPyV archetype isolates, short deletions or duplications were observed, corroborating that this region is highly unstable [66,67]. Therefore, it is possible to assume that subsequent archetypal NCRR rearrangements could determine the onset of PML strains, such as Mad-1 [68].
Based on the occurrence in the NCRR of transcriptional enhancer repeat elements, BKPyV isolates can also be identified as archetype and prototype strains. The archetype BKPyV WW strain, characterized by five blocks named O (35 bp), which includes the origin of replication and a TATA-box, P (68 bp), Q (39 bp), R (63 bp), and S (63 bp), containing TATA-like elements and the regulatory regions for early and late genes expression, is considered the infectious strain, shed in the urine of immunocompetent individuals [69–71]. Approximately 30 transcription factor binding sites are in silico predicted: SP1 has been the most extensively studied [72–74], although the additional role played by other transcription factors such as NF1, ETS1, NFκB, the glucocorticoid and progesterone receptors, and CREB were evidenced in several studies [73,75–77].
Similarly to JCPyV, the plausible instability of the archetype BKPyV NCRR could contribute to the development of the prototype strains, which is able to cause polyomavirus-associated nephropathy in kidney transplant recipients and hemorrhagic cystitis in hematopoietic stem cell transplant recipients [78–81]. The Dunlop strain, the most salient prototype strain, was isolated from a kidney transplant recipient with ureteral stenosis [82]. This strain displays three 68-bp tandem repeat within the NCRR (O-P-P-P-S arrangement) with respect to the archetype strain, carrying a single 68-bp motif. This strain showed less enhancer activity than the prototype strain, thus confirming the significance of the triplicated motifs on transcriptional regulation, and on viral infectious activity [83]. In fact, BKPyV strains isolated from kidney transplant recipients with rearranged NCRR showed higher viral gene expression and viral loads with more extensive pathogenicity [84].
Additional NCCR structures have been described for both viruses [85–88]. In particular, the presence of a common pattern of JCPyV NCCR rearrangement, such as the D-box deletion, can be considered a hallmark needed for the initial NCCR rearrangements critical co-factor for the development of PML in immunosuppressed individuals [88,89]. Besides the triplication of the P region, rearrangements of BKPyV NCCR involve the adjacent O and Q blocks. Differently, the S block is always retained, hence highlighting the importance of these nucleotide sequences [70]. NCCR mutations were also observed during in vitro JCPyV and BKPyV cultivation, confirming that NCRR variants could arise after prolonged propagation of the viruses in cells [71,85,90,91]. The mechanisms by which both viruses determine relevant human diseases are not established, but it is accepted that the regulation of gene expression in HPyVs plays a role in determining the viral tropism, and in the promotion of pathogenesis progression [92].
Little is known about the genetic diversity of the NCCRs from the novel HPyVs and the biological relevance in terms of viral transcription, replication, and possible pathogenic properties. In this review, we provide an overview of the mutations in the NCCR, which is defined as the sequence between the start codon of the LT/sT gene and the start codon of the VP2 gene, of the novel HPyVs and their known effect on promoter activity. We discuss how NCCR rearrangements may affect the binding of putative transcription factors, and whether specific NCCR configurations are associated with disease.
This entry is adapted from the peer-reviewed paper 10.3390/v12121406