Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Hematological abnormalities are frequently linked to chronic liver disease of any etiology. About 75% of patients with advanced chronic liver disease experience anemia. The causes of anemia are complex and multifactorial, particularly in cirrhotic patients.
- chronic liver disease
- anemia
- hepcidin
1. Introduction
Anemias represent a category of diseases defined by a decrease in the blood concentration of hemoglobin (Hb) below 120 g/L in females and 130 g/L in males.
Transporting CO2 from the tissues to the lungs and oxygen from the lungs to the tissues is the job of erythrocytes. This is done by Hb (a tetrameric protein) consisting of heme and globin. In anemias, the ability to transport O2 by erythrocytes is altered, and the compensation mechanisms aim to improve the transport of O2 in the blood.
Following the scheme of the evolution of the red series, we can establish the pathogenesis of each type of anemia: damage to the pluripotent stem cells and erythrocyte precursors (“erythroid burst colony-forming unit”) will lead to hypoproliferative anemias (aplastic-hypoplastic), a disorder in DNA synthesis that is responsible for the constitution of megaloblastic anemias.
A disturbance in Hb synthesis leads to hypochromic anemias; acute and chronic hemorrhages characterize posthemorrhagic anemias; premature destruction of erythrocytes is characteristic of hemolytic anemias.
The pathogenetic classification of anemias is the most important and valuable in practice, secondary to erythrocyte morphology.
Between 50% and 87% of patients with advanced chronic liver disease (ACLD) have anemia, according to a number of observational studies, with patients with liver encephalopathy having the highest prevalence [1,2,3,4].
Several etiological factors of anemia in patients with ACLD have been described (Figure 1): acute or chronic blood loss due to gastroesophageal varices bleeding [5], portal hypertensive gastropathy, gastric antral vascular ectasia (GAVE) [6], or peptic ulcer [7,8], which aggravates anemia in ACLD. Defects of the lipid membrane of erythrocytes (functional and structural) can lead to the formation of acanthocytes, which have a short life, due to an increased susceptibility to degradation in the spleen [9,10].
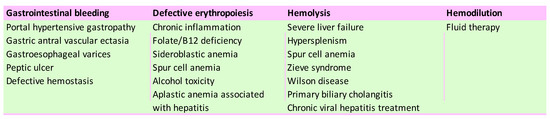
Figure 1. Leading causes of anemia in chronic liver disease patients.
2. Role of Iron Deficiency in Chronic Liver Diseases
Iron homeostasis is significantly influenced by the liver (Figure 2). Hepcidin, a hormone that regulates iron and is expressed in situations of excess iron and inflammation, is primarily produced by the liver. The contribution of hepcidin in the pathogenesis of liver diseases is still being investigated. Hepcidin represents one of the factors contributing to anemia in chronic liver diseases because it blocks iron absorption from enterocytes [17].
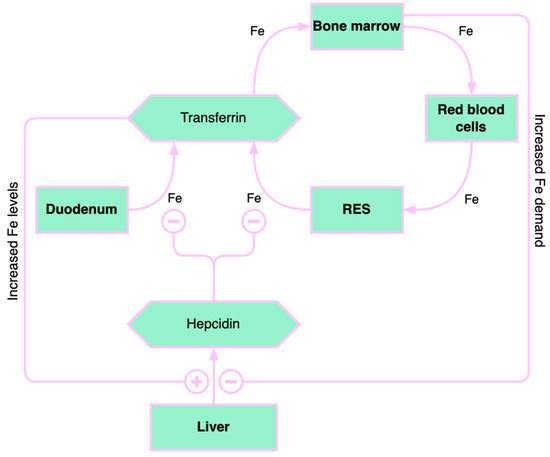
Figure 2. Overview of iron homeostasis. RES: reticuloendothelial system. “+” means “increase” and “–” means “decrease”.
Low hepcidin levels were found in chronic liver disease (CLD), especially in patients with severe liver injury. In addition, CLDs (alcoholic and non-alcoholic liver disease and chronic viral hepatitis C) are linked to varying degrees of iron overload, which, in addition to inflammation, is a complex, but not fully understood, prerequisite for the production and control of hepcidin [17].
Genetic disorders of iron overload and iron deficiency, such as hereditary hemochromatosis and iron-refractory anemia, are characterized by abnormalities in the regulation of hepcidin production. In ineffective erythropoiesis, excessive suppression of hepcidin causes iron overload anemia (e.g., β-thalassemia), and hepcidin excess contributes to iron deficiency anemia and anemia associated with chronic inflammatory diseases (ACD) [18]. Tsochatzis et al. reported a close relationship between low hepcidin levels and the degree of liver inflammation and biochemical and histological inflammation markers, hepatocellular injury, and fibrosis, especially in patients with chronic viral hepatitis C [19].
According to a recent study, patients with autoimmune hepatitis, primary biliary cholangitis, and primary sclerosing cholangitis had lower serum hepcidin levels and a lower hepcidin/ferritin ratio than patients with chronic viral hepatitis B and C [20].
In contrast, Wang et al. noted a higher mean hepcidin level in patients with chronic hepatitis B without cirrhosis as well as in patients with hepatocellular carcinoma compared to the healthy population, but not in the cirrhotic patients, in an observational study on 46 patients with chronic hepatitis B whether or not diagnosed with liver cirrhosis. This information implied that, in addition to liver inflammation, iron loading and viral infection may also control hepcidin in the non-cirrhotic group [21].
Decreased hepcidin levels in cirrhotic patients cause hepatic iron overload, with the secondary progression of liver fibrosis [22].
Extensive and comprehensive studies of pathogenetic mechanisms of hepcidin regulation postulated that hepcidin plays an essential role in iron hemostasis [23], future research being necessary to elucidate the existence of other factors that modulate its expression in liver diseases of various etiologies.
2.1. Liver Iron Storage and Chronic Liver Disease
The most important indicator of iron homeostasis is ferritin. The hepatocyte is the leading site for the synthesis of ferritin, as well as for the synthesis of transferrin, which is the main iron-binding protein. In CLD, an imbalance between iron deficiency and iron overload has been reported. Most of the iron amount is stored in ferritin, but hemosiderin is also used in case of severe iron overload [24]. Hepatic iron overload is associated with a worse outcome and with an increased risk of hepatocarcinoma [25].
In patients with severe CLD, there were described reduced levels of serum transferrin and iron and increased serum ferritin levels correlated with higher values of serum liver enzymes, which suggested the alleged role of inflammation in the pathogenesis of anemia in these patients [26].
In chronic inflammatory conditions, including chronic viral hepatitis, autoimmune hepatitis, chronic kidney and heart failure, organ rejection, inflammatory bowel diseases, endocrine diseases, systemic autoimmune diseases, and neoplasms, the impairment of erythropoiesis is secondary to an altered iron hemostasis, a cytokine-mediated process [27]. Proinflammatory cytokines, such as tumor necrosis factor (TNF-α) and interleukin-1, 6, and 10, are the main factors limiting iron availability in erythrocyte precursor cells, with a consequent alteration of erythropoiesis both in nonalcoholic steatohepatitis (NASH) and in chronic viral hepatitis [28].
In NASH, an increased expression of proinflammatory cytokines and hepcidin was described by Handa et al., who associated obesity-induced inflammation with increased serum hepcidin, which induces ACD [29].
ACD, also referred to as inflammatory anemia, is initially characterized by mild to moderate normochromic normocytic anemia and features of iron deficiency anemia (IDA), such as decreased reticulocytes and transferrin saturation. Anemia in CLD is primarily determined by IDA and ACD [30].
2.2. The Causes of Iron Deficiency in Chronic Liver Diseases
Patients with liver cirrhosis present a higher risk of gastrointestinal bleeding due to the decreased production of coagulation factors and thrombocytopenia, which, correlated with the degree of portal hypertension, defines the risk of bleeding in the gastrointestinal tract [31].
The most common cause of ID in advanced CLD is chronic gastrointestinal bleeding. ID causes IDA, a common pathology in this population [32,33]. Acute bleeding usually occurs through the rupture of esophageal varices or a hemorrhagic peptic ulcer [34]. Chronic blood loss is correlated with portal hypertensive gastropathy (PHG) and gastric antral vascular ectasia (GAVE), two primary gastric mucosa lesions caused by vascular ectasia. Histologically, it is defined by vascular dilatation in the mucosa and submucosa of the gastric body and fundus, without significant inflammation. The severity of PHG correlates to the stage of portal hypertension and the blood flow of the gastric mucosa or other local factors, which may determine its pathogenesis [35]. GAVE (“watermelon stomach”) is a rare condition that causes about 4% of non-variceal gastrointestinal bleeding [36]. Endoscopically, it is characterized by vascular ectasia with a sinuous appearance at the level of the longitudinal folds of the gastric antrum. The formation of GAVE is influenced by the presence of liver failure, which contributes to its pathogenesis, GAVE being more common in severe liver diseases. Other diseases besides liver failure can also cause the development of a “watermelon stomach”, such as chronic kidney injury, connective tissue diseases, or bone marrow transplantation. The pathogenesis is unclear; vasodilator agents such as prostaglandin E2 are not metabolized by the liver. Mechanical stress and hypomobility of the stomach could be other factors associated with GAVE [37].
The most common etiologies of lower gastrointestinal bleeding in liver cirrhosis are hemorrhoidal disease and portal hypertensive colopathy. Portal hypertensive enteropathy may be an occult source of chronic gastrointestinal bleeding in liver cirrhosis [38].
3. Pathophysiological Mechanisms of Anemia in Alcoholic Liver Disease
Alcohol consumption contributes to anemia through multiple mechanisms (Figure 3). Alcohol directly affects the hematopoietic cells of the bone marrow, manifesting as pancytopenia, reversible after stopping alcohol consumption. Folate deficiency, found in these patients, leads to macrocytic anemia. Alcohol decreases serum folate levels through various mechanisms, such as increased urinary folate excretion and decreased jejunal absorption [39]. Through its effects on methionine metabolism, folate deficiency contributes to the progression of liver disease by influencing DNA synthesis and stability as well as the epigenetic regulation of gene expression that is implicated in causing liver damage [40].
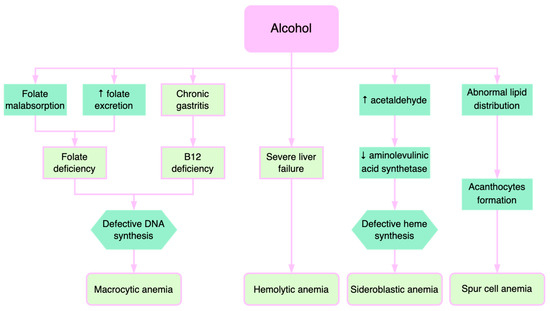
Figure 3. Pathogenetic mechanisms of alcohol-induced anemia.
Patients with severe liver injury exhibit deficiencies of folic acid and vitamin B12, causing macrocytic anemia. Vitamin B12 and folate are necessary for the synthesis of thymidylate and purines. Therefore, their deficiencies lead to defects in DNA synthesis with subsequent macrocytic anemia [41]. Additionally, increased cholesterol deposition on the erythrocyte membrane, which in turn raises the erythrocyte surface, may contribute to macrocytic anemia in liver diseases [41,42].
Severe liver failure is also correlated with hemolytic anemias, where excessive destruction of erythrocytes and increased reticulocytes are described [43]. Immature erythrocytes are about 20% larger than mature erythrocytes, which explains macrocytosis.
Acetaldehyde, the product resulting from alcohol oxidation in the liver, increases the degradation of pyridoxal phosphate, which is a cofactor of aminolevulinic acid synthetase, an enzyme with a catalytic role in heme synthesis. Consequently, alcohol causes the inadequate production of heme and consequently causes the accumulation of iron in the mitochondria, determining the formation of sideroblasts [44]. Sideroblastic anemia is encountered in approximately 25–30% of alcoholic patients, reversible when consumption is stopped. Sideroblastic anemia occurs in alcoholic patients with concomitant folic acid deficiency [45].
The formation of acanthocytes (spur cells) is another mechanism of anemia in alcoholic liver disease [46]. The abnormal lipid metabolism leads to defects in lipid membrane composition and consequent formation of acanthocytes. Spur cell anemia (SCA) is present in up to 31% of patients with advanced liver cirrhosis, an independent negative predictive factor associated with an increased mortality risk [10,47]. SCA prognosis is extremely poor, with liver transplantation being the only effective treatment [48,49].
This entry is adapted from the peer-reviewed paper 10.3390/gastroent14030024
This entry is offline, you can click here to edit this entry!