Ewing sarcoma (EwS) is a highly aggressive and metastatic cancer in children and adolescents. Canonical therapy mainly comprises the combination of intensive chemotherapy, radiation, and local surgery, which give rise to acute and chronic adverse effects. Drugs targeting EwS without side effects are in urgent demand. Genetically, EwS is characterized by chromosomal translocations with a low mutation burden. As a result, the chimeric protein EWS-ETS, mainly EWS-FLI1(85%), is critical for the malignancy of EwS. EWS-FLI1 directly binds to GGAA microsatellites in enhancers and promotors of the target genes and recruits multiple transcription factors or epigenetic regulators to reprogramme the epigenome.
- ewing sarcoma
- EWS-FLI1
- protein complex
- targeted therapy
1. Introduction
2. Involvement of EWS-FLI1 in Transcription, Epigenetic Reprogramming, and Alternative Splicing in EwS
2.1. EWS-FLI1 in Transcription and Epigenetic Reprogramming
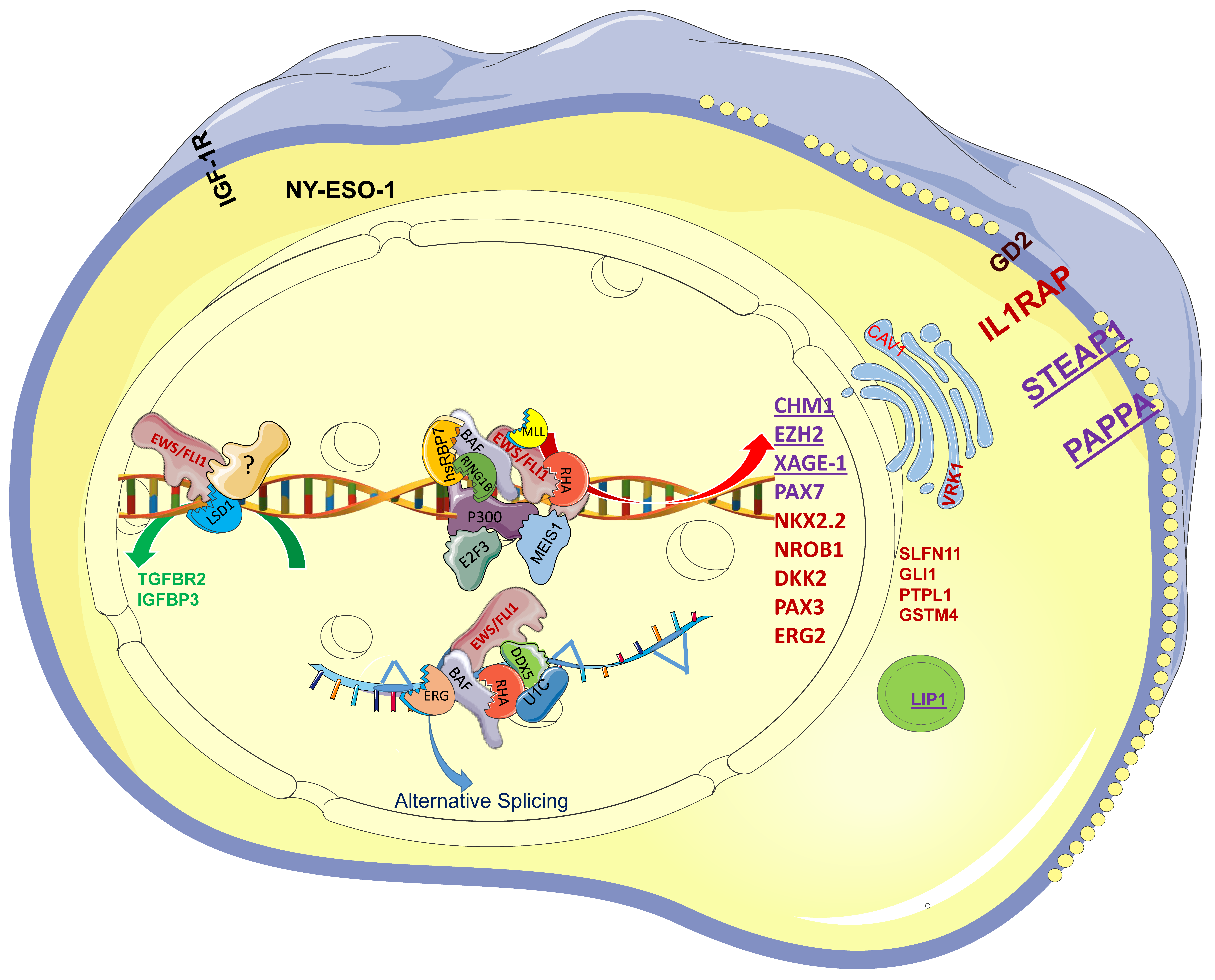
2.2. EWS-FLI1 in Alternative Splicing
3. The Regulation of EWS-FLI1
4. CAR-T Therapy
This entry is adapted from the peer-reviewed paper 10.3390/cancers15164035
References
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5.
- Jahanseir, K.; Folpe, A.L.; Graham, R.P.; Giannini, C.; Robinson, S.I.; Sukov, W.; Fritchie, K. Ewing Sarcoma in Older Adults: A Clinicopathologic Study of 50 Cases Occurring in Patients Aged >/=40 Years, With Emphasis on Histologic Mimics. Int. J. Surg. Pathol. 2020, 28, 352–360.
- Riggi, N.; Suva, M.L.; Stamenkovic, I. Ewing’s sarcoma origin: From duel to duality. Expert Rev. Anticancer Ther. 2009, 9, 1025–1030.
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429.
- Kovar, H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin. Ther. Targets 2014, 18, 1315–1328.
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046.
- Haeusler, J.; Ranft, A.; Boelling, T.; Gosheger, G.; Braun-Munzinger, G.; Vieth, V.; Burdach, S.; van den Berg, H.; Juergens, H.; Dirksen, U. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010, 116, 443–450.
- Zollner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Alava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685.
- Ladenstein, R.; Potschger, U.; Le Deley, M.C.; Whelan, J.; Paulussen, M.; Oberlin, O.; van den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. J. Clin. Oncol. 2010, 28, 3284–3291.
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e119.
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681.
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 2014, 20, 4584–4597.
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773.
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095.
- Richter, G.H.S.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304.
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schonegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395.
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329.
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154.
- Gangwal, K.; Lessnick, S.L. Microsatellites are EWS/FLI response elements: Genomic “junk” is EWS/FLI’s treasure. Cell Cycle 2008, 7, 3127–3132.
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE 2009, 4, e4932.
- Yang, L.; Chansky, H.A.; Hickstein, D.D. EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J. Biol. Chem. 2000, 275, 37612–37618.
- Ahmed, N.S.; Harrell, L.M.; Wieland, D.R.; Lay, M.A.; Thompson, V.F.; Schwartz, J.C. Fusion protein EWS-FLI1 is incorporated into a protein granule in cells. RNA 2021, 27, 920–932.
- Petermann, R.; Mossier, B.M.; Aryee, D.N.; Khazak, V.; Golemis, E.A.; Kovar, H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene 1998, 17, 603–610.
- Zhou, H.; Lee, K.A. An hsRPB4/7-dependent yeast assay for trans-activation by the EWS oncogene. Oncogene 2001, 20, 1519–1524.
- Bilke, S.; Schwentner, R.; Yang, F.; Kauer, M.; Jug, G.; Walker, R.L.; Davis, S.; Zhu, Y.J.; Pineda, M.; Meltzer, P.S.; et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013, 23, 1797–1809.
- Schwentner, R.; Papamarkou, T.; Kauer, M.O.; Stathopoulos, V.; Yang, F.; Bilke, S.; Meltzer, P.S.; Girolami, M.; Kovar, H. EWS-FLI1 employs an E2F switch to drive target gene expression. Nucleic Acids Res. 2015, 43, 2780–2789.
- Mertens, F.; Antonescu, C.R.; Mitelman, F. Gene fusions in soft tissue tumors: Recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016, 55, 291–310.
- Ramakrishnan, R.; Fujimura, Y.; Zou, J.P.; Liu, F.; Lee, L.; Rao, V.N.; Reddy, E.S. Role of protein-protein interactions in the antiapoptotic function of EWS-Fli-1. Oncogene 2004, 23, 7087–7094.
- Harlow, M.L.; Chasse, M.H.; Boguslawski, E.A.; Sorensen, K.M.; Gedminas, J.M.; Kitchen-Goosen, S.M.; Rothbart, S.B.; Taslim, C.; Lessnick, S.L.; Peck, A.S.; et al. Trabectedin Inhibits EWS-FLI1 and Evicts SWI/SNF from Chromatin in a Schedule-dependent Manner. Clin. Cancer Res. 2019, 25, 3417–3429.
- Monument, M.J.; Johnson, K.M.; McIlvaine, E.; Abegglen, L.; Watkins, W.S.; Jorde, L.B.; Womer, R.B.; Beeler, N.; Monovich, L.; Lawlor, E.R.; et al. Clinical and biochemical function of polymorphic NR0B1 GGAA-microsatellites in Ewing sarcoma: A report from the Children’s Oncology Group. PLoS ONE 2014, 9, e104378.
- Lin, L.; Huang, M.; Shi, X.; Mayakonda, A.; Hu, K.; Jiang, Y.Y.; Guo, X.; Chen, L.; Pang, B.; Doan, N.; et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic Acids Res. 2019, 47, 1255–1267.
- Sanchez-Molina, S.; Figuerola-Bou, E.; Blanco, E.; Sanchez-Jimenez, M.; Taboas, P.; Gomez, S.; Ballare, C.; Garcia-Dominguez, D.J.; Prada, E.; Hontecillas-Prieto, L.; et al. RING1B recruits EWSR1-FLI1 and cooperates in the remodeling of chromatin necessary for Ewing sarcoma tumorigenesis. Sci. Adv. 2020, 6.
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416.
- Kinsey, M.; Smith, R.; Lessnick, S.L. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol. Cancer Res. 2006, 4, 851–859.
- Boro, A.; Pretre, K.; Rechfeld, F.; Thalhammer, V.; Oesch, S.; Wachtel, M.; Schafer, B.W.; Niggli, F.K. Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Int. J. Cancer 2012, 131, 2153–2164.
- Garcia-Aragoncillo, E.; Carrillo, J.; Lalli, E.; Agra, N.; Gomez-Lopez, G.; Pestana, A.; Alonso, J. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene 2008, 27, 6034–6043.
- Cironi, L.; Riggi, N.; Provero, P.; Wolf, N.; Suva, M.L.; Suva, D.; Kindler, V.; Stamenkovic, I. IGF1 is a common target gene of Ewing’s sarcoma fusion proteins in mesenchymal progenitor cells. PLoS ONE 2008, 3, e2634.
- Wiles, E.T.; Lui-Sargent, B.; Bell, R.; Lessnick, S.L. BCL11B is up-regulated by EWS/FLI and contributes to the transformed phenotype in Ewing sarcoma. PLoS ONE 2013, 8, e59369.
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082.
- Abaan, O.D.; Levenson, A.; Khan, O.; Furth, P.A.; Uren, A.; Toretsky, J.A. PTPL1 is a direct transcriptional target of EWS-FLI1 and modulates Ewing’s Sarcoma tumorigenesis. Oncogene 2005, 24, 2715–2722.
- Luo, W.; Xu, C.; Ayello, J.; Dela Cruz, F.; Rosenblum, J.M.; Lessnick, S.L.; Cairo, M.S. Protein phosphatase 1 regulatory subunit 1A in ewing sarcoma tumorigenesis and metastasis. Oncogene 2018, 37, 798–809.
- Grunewald, T.G.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet. 2015, 47, 1073–1078.
- Luo, W.; Gangwal, K.; Sankar, S.; Boucher, K.M.; Thomas, D.; Lessnick, S.L. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 2009, 28, 4126–4132.
- Charville, G.W.; Wang, W.L.; Ingram, D.R.; Roy, A.; Thomas, D.; Patel, R.M.; Hornick, J.L.; van de Rijn, M.; Lazar, A.J. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod. Pathol. 2017, 30, 1312–1320.
- Von Heyking, K.; Calzada-Wack, J.; Gollner, S.; Neff, F.; Schmidt, O.; Hensel, T.; Schirmer, D.; Fasan, A.; Esposito, I.; Muller-Tidow, C.; et al. The endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcoma. Mol. Oncol. 2017, 11, 1288–1301.
- Zhou, Z.; Yu, L.; Kleinerman, E.S. EWS-FLI-1 regulates the neuronal repressor gene REST, which controls Ewing sarcoma growth and vascular morphology. Cancer 2014, 120, 579–588.
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Muller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65.
- Grunewald, T.G.P.; Ranft, A.; Esposito, I.; da Silva-Buttkus, P.; Aichler, M.; Baumhoer, D.; Schaefer, K.L.; Ottaviano, L.; Poremba, C.; Jundt, G.; et al. High STEAP1 expression is associated with improved outcome of Ewing’s sarcoma patients. Ann. Oncol. 2012, 23, 2185–2190.
- Tang, S.W.; Bilke, S.; Cao, L.; Murai, J.; Sousa, F.G.; Yamade, M.; Rajapakse, V.; Varma, S.; Helman, L.J.; Khan, J.; et al. SLFN11 Is a Transcriptional Target of EWS-FLI1 and a Determinant of Drug Response in Ewing Sarcoma. Clin. Cancer Res. 2015, 21, 4184–4193.
- Ma, Y.; Baltezor, M.; Rajewski, L.; Crow, J.; Samuel, G.; Staggs, V.S.; Chastain, K.M.; Toretsky, J.A.; Weir, S.J.; Godwin, A.K. Targeted inhibition of histone deacetylase leads to suppression of Ewing sarcoma tumor growth through an unappreciated EWS-FLI1/HDAC3/HSP90 signaling axis. J. Mol. Med. 2019, 97, 957–972.
- He, S.; Huang, Q.; Hu, J.; Li, L.; Xiao, Y.; Yu, H.; Han, Z.; Wang, T.; Zhou, W.; Wei, H.; et al. EWS-FLI1-mediated tenascin-C expression promotes tumour progression by targeting MALAT1 through integrin alpha5beta1-mediated YAP activation in Ewing sarcoma. Br. J. Cancer 2019, 121, 922–933.
- Zhang, H.F.; Hughes, C.S.; Li, W.; He, J.Z.; Surdez, D.; El-Naggar, A.M.; Cheng, H.; Prudova, A.; Delaidelli, A.; Negri, G.L.; et al. Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discov. 2021, 11, 2884–2903.
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978.
- Bailly, R.A.; Bosselut, R.; Zucman, J.; Cormier, F.; Delattre, O.; Roussel, M.; Thomas, G.; Ghysdael, J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol. Cell. Biol. 1994, 14, 3230–3241.
- Li, J.; Ohmura, S.; Marchetto, A.; Orth, M.F.; Imle, R.; Dallmayer, M.; Musa, J.; Knott, M.M.L.; Holting, T.L.B.; Stein, S.; et al. Therapeutic targeting of the PLK1-PRC1-axis triggers cell death in genomically silent childhood cancer. Nat. Commun. 2021, 12, 5356.
- Owen, L.A.; Kowalewski, A.A.; Lessnick, S.L. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS ONE 2008, 3, e1965.
- Shi, X.; Zheng, Y.; Jiang, L.; Zhou, B.; Yang, W.; Li, L.; Ding, L.; Huang, M.; Gery, S.; Lin, D.C.; et al. EWS-FLI1 regulates and cooperates with core regulatory circuitry in Ewing sarcoma. Nucleic Acids Res. 2020, 48, 11434–11451.
- Markey, F.B.; Romero, B.; Parashar, V.; Batish, M. Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells 2021, 10, 1300.
- Kinsey, M.; Smith, R.; Iyer, A.K.; McCabe, E.R.; Lessnick, S.L. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009, 69, 9047–9055.
- Tang, S.W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953.
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell. Biol. 2004, 24, 7275–7283.
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.J.; Lessnick, S.L. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2013, 32, 5089–5100.
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304.
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596.
- Agra, N.; Cidre, F.; Garcia-Garcia, L.; de la Parra, J.; Alonso, J. Lysyl oxidase is downregulated by the EWS/FLI1 oncoprotein and its propeptide domain displays tumor supressor activities in Ewing sarcoma cells. PLoS ONE 2013, 8, e66281.
- Katschnig, A.M.; Kauer, M.O.; Schwentner, R.; Tomazou, E.M.; Mutz, C.N.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.N.T.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017, 36, 5995–6005.
- Niedan, S.; Kauer, M.; Aryee, D.N.; Kofler, R.; Schwentner, R.; Meier, A.; Potschger, U.; Kontny, U.; Kovar, H. Suppression of FOXO1 is responsible for a growth regulatory repressive transcriptional sub-signature of EWS-FLI1 in Ewing sarcoma. Oncogene 2014, 33, 3927–3938.
- Dylla, L.; Moore, C.; Jedlicka, P. MicroRNAs in Ewing Sarcoma. Front. Oncol. 2013, 3, 65.
- Riggi, N.; Suva, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suva, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932.
- Robin, T.P.; Smith, A.; McKinsey, E.; Reaves, L.; Jedlicka, P.; Ford, H.L. EWS/FLI1 regulates EYA3 in Ewing sarcoma via modulation of miRNA-708, resulting in increased cell survival and chemoresistance. Mol. Cancer Res. 2012, 10, 1098–1108.
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856.
- Nakatani, F.; Ferracin, M.; Manara, M.C.; Ventura, S.; Del Monaco, V.; Ferrari, S.; Alberghini, M.; Grilli, A.; Knuutila, S.; Schaefer, K.L.; et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J. Pathol. 2012, 226, 796–805.
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191.
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016.
- Wang, J.; Lee, J.E.; Riemondy, K.; Yu, Y.; Marquez, S.M.; Lai, E.C.; Yi, R. XPO5 promotes primary miRNA processing independently of RanGTP. Nat. Commun. 2020, 11, 1845.
- Selvanathan, S.P.; Graham, G.T.; Erkizan, H.V.; Dirksen, U.; Natarajan, T.G.; Dakic, A.; Yu, S.; Liu, X.; Paulsen, M.T.; Ljungman, M.E.; et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc. Natl. Acad. Sci. USA 2015, 112, E1307–E1316.
- Sun, H.L.; Cui, R.; Zhou, J.; Teng, K.Y.; Hsiao, Y.H.; Nakanishi, K.; Fassan, M.; Luo, Z.; Shi, G.; Tili, E.; et al. ERK Activation Globally Downregulates miRNAs through Phosphorylating Exportin-5. Cancer Cell 2016, 30, 723–736.
- Klevernic, I.V.; Morton, S.; Davis, R.J.; Cohen, P. Phosphorylation of Ewing’s sarcoma protein (EWS) and EWS-Fli1 in response to DNA damage. Biochem. J. 2009, 418, 625–634.
- Bachmaier, R.; Aryee, D.N.; Jug, G.; Kauer, M.; Kreppel, M.; Lee, K.A.; Kovar, H. O-GlcNAcylation is involved in the transcriptional activity of EWS-FLI1 in Ewing’s sarcoma. Oncogene 2009, 28, 1280–1284.
- Olsen, R.J.; Hinrichs, S.H. Phosphorylation of the EWS IQ domain regulates transcriptional activity of the EWS/ATF1 and EWS/FLI1 fusion proteins. Oncogene 2001, 20, 1756–1764.
- Schlottmann, S.; Erkizan, H.V.; Barber-Rotenberg, J.S.; Knights, C.; Cheema, A.; Uren, A.; Avantaggiati, M.L.; Toretsky, J.A. Acetylation Increases EWS-FLI1 DNA Binding and Transcriptional Activity. Front. Oncol. 2012, 2, 107.
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243.
- Eymin, B. Targeting the spliceosome machinery: A new therapeutic axis in cancer? Biochem. Pharmacol. 2021, 189, 114039.
- Embree, L.J.; Azuma, M.; Hickstein, D.D. Ewing sarcoma fusion protein EWSR1/FLI1 interacts with EWSR1 leading to mitotic defects in zebrafish embryos and human cell lines. Cancer Res. 2009, 69, 4363–4371.
- Knoop, L.L.; Baker, S.J. The splicing factor U1C represses EWS/FLI-mediated transactivation. J. Biol. Chem. 2000, 275, 24865–24871.
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636.
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schafer, B.W.; Burdach, S.; Richter, G.H. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2016, 7, 1451–1463.
- García-Domínguez, D.J.; Hajji, N.; Sánchez-Molina, S.; Figuerola-Bou, E.; de Pablos, R.M.; Espinosa-Oliva, A.M.; Andrés-León, E.; Terrón-Camero, L.C.; Flores-Campos, R.; Pascual-Pasto, G.; et al. HDAC6 regulates expression of the oncogenic driver EWSR1-FLI1 through the <em>EWSR1</em> promoter in Ewing sarcoma. bioRxiv 2021.
- Ban, J.; Jug, G.; Mestdagh, P.; Schwentner, R.; Kauer, M.; Aryee, D.N.; Schaefer, K.L.; Nakatani, F.; Scotlandi, K.; Reiter, M.; et al. Hsa-mir-145 is the top EWS-FLI1-repressed microRNA involved in a positive feedback loop in Ewing’s sarcoma. Oncogene 2011, 30, 2173–2180.
- Keskin, T.; Bakaric, A.; Waszyk, P.; Boulay, G.; Torsello, M.; Cornaz-Buros, S.; Chevalier, N.; Geiser, T.; Martin, P.; Volorio, A.; et al. LIN28B Underlies the Pathogenesis of a Subclass of Ewing Sarcoma LIN28B Control of EWS-FLI1 Stability. Cell Rep. 2020, 30, 4567–4583.e5.
- De Vito, C.; Riggi, N.; Suva, M.L.; Janiszewska, M.; Horlbeck, J.; Baumer, K.; Provero, P.; Stamenkovic, I. Let-7a is a direct EWS-FLI-1 target implicated in Ewing’s sarcoma development. PLoS ONE 2011, 6, e23592.
- Gierisch, M.E.; Pfistner, F.; Lopez-Garcia, L.A.; Harder, L.; Schafer, B.W.; Niggli, F.K. Proteasomal Degradation of the EWS-FLI1 Fusion Protein Is Regulated by a Single Lysine Residue. J. Biol. Chem. 2016, 291, 26922–26933.
- Gierisch, M.E.; Pedot, G.; Walser, F.; Lopez-Garcia, L.A.; Jaaks, P.; Niggli, F.K.; Schafer, B.W. USP19 deubiquitinates EWS-FLI1 to regulate Ewing sarcoma growth. Sci. Rep. 2019, 9, 951.
- Seong, B.K.A.; Dharia, N.V.; Lin, S.; Donovan, K.A.; Chong, S.; Robichaud, A.; Conway, A.; Hamze, A.; Ross, L.; Alexe, G.; et al. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell 2021, 39, 1262–1278.e1267.
- Su, S.; Chen, J.; Jiang, Y.; Wang, Y.; Vital, T.; Zhang, J.; Laggner, C.; Nguyen, K.T.; Zhu, Z.; Prevatte, A.W.; et al. SPOP and OTUD7A Control EWS-FLI1 Protein Stability to Govern Ewing Sarcoma Growth. Adv. Sci. 2021, 8, e2004846.
- Sun, H.; Lin, D.C.; Cao, Q.; Guo, X.; Marijon, H.; Zhao, Z.; Gery, S.; Xu, L.; Yang, H.; Pang, B.; et al. CRM1 Inhibition Promotes Cytotoxicity in Ewing Sarcoma Cells by Repressing EWS-FLI1-Dependent IGF-1 Signaling. Cancer Res. 2016, 76, 2687–2697.
- Bailey, K.M.; Julian, C.M.; Klinghoffer, A.N.; Bernard, H.; Lucas, P.C.; McAllister-Lucas, L.M. EWS-FLI1 low Ewing sarcoma cells demonstrate decreased susceptibility to T-cell-mediated tumor cell apoptosis. Oncotarget 2019, 10, 3385–3399.
- Sengupta, A.; Rahman, M.; Mateo-Lozano, S.; Tirado, O.M.; Notario, V. The dual inhibitory effect of thiostrepton on FoxM1 and EWS/FLI1 provides a novel therapeutic option for Ewing’s sarcoma. Int. J. Oncol. 2013, 43, 803–812.
- Stegmaier, K.; Wong, J.S.; Ross, K.N.; Chow, K.T.; Peck, D.; Wright, R.D.; Lessnick, S.L.; Kung, A.L.; Golub, T.R. Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med. 2007, 4, e122.
- DuBois, S.G.; Krailo, M.D.; Lessnick, S.L.; Smith, R.; Chen, Z.; Marina, N.; Grier, H.E.; Stegmaier, K.; Children’s Oncology, G. Phase II study of intermediate-dose cytarabine in patients with relapsed or refractory Ewing sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2009, 52, 324–327.
- Chu, Z.; Gu, L.; Hu, Y.; Zhang, X.; Li, M.; Chen, J.; Teng, D.; Huang, M.; Shen, C.H.; Cai, L.; et al. STAG2 regulates interferon signaling in melanoma via enhancer loop reprogramming. Nat. Commun. 2022, 13, 1859.
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353.
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035.
- Adane, B.; Alexe, G.; Seong, B.K.A.; Lu, D.; Hwang, E.E.; Hnisz, D.; Lareau, C.A.; Ross, L.; Lin, S.; Dela Cruz, F.S.; et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell 2021, 39, 827–844.e810.
- Surdez, D.; Zaidi, S.; Grossetete, S.; Laud-Duval, K.; Ferre, A.S.; Mous, L.; Vourc’h, T.; Tirode, F.; Pierron, G.; Raynal, V.; et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021, 39, 810–826.e819.
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341.
- Lu, D.Y.; Ellegast, J.M.; Ross, K.N.; Malone, C.F.; Lin, S.; Mabe, N.W.; Dharia, N.V.; Meyer, A.; Conway, A.; Su, A.H.; et al. The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma. Nat. Cell Biol. 2023, 25, 285–297.
- Gao, Y.; He, X.Y.; Wu, X.S.; Huang, Y.H.; Toneyan, S.; Ha, T.; Ipsaro, J.J.; Koo, P.K.; Joshua-Tor, L.; Bailey, K.M.; et al. ETV6 dependency in Ewing sarcoma by antagonism of EWS-FLI1-mediated enhancer activation. Nat. Cell Biol. 2023, 25, 298–308.
- Berghuis, D.; de Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231.
- Morales, E.; Olson, M.; Iglesias, F.; Dahiya, S.; Luetkens, T.; Atanackovic, D. Role of immunotherapy in Ewing sarcoma. J. Immunother. Cancer 2020, 8, e000653.
- Englisch, A.; Altvater, B.; Kailayangiri, S.; Hartmann, W.; Rossig, C. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr. Blood Cancer 2020, 67, e28313.
- Challita-Eid, P.M.; Morrison, K.; Etessami, S.; An, Z.; Morrison, K.J.; Perez-Villar, J.J.; Raitano, A.B.; Jia, X.C.; Gudas, J.M.; Kanner, S.B.; et al. Monoclonal antibodies to six-transmembrane epithelial antigen of the prostate-1 inhibit intercellular communication in vitro and growth of human tumor xenografts in vivo. Cancer Res. 2007, 67, 5798–5805.
- Richter, G.H.; Fasan, A.; Hauer, K.; Grunewald, T.G.; Berns, C.; Rossler, S.; Naumann, I.; Staege, M.S.; Fulda, S.; Esposito, I.; et al. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J. Pathol. 2013, 230, 70–81.
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152.
- Schirmer, D.; Storz, I.; Wisskirchen, K.; Feederle, R.; Schmidt, O.; Abken, H.; Protzer, U.; Burdach, S.; Richter, G.H.S. GPR64-specific CAR-transgenic T cells selectively kill Ewing sarcoma in vivo. Oncol. Res. Treat. 2018, 41, 130–131.
- Town, J.; Pais, H.; Harrison, S.; Stead, L.F.; Bataille, C.; Bunjobpol, W.; Zhang, J.; Rabbitts, T.H. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl. Acad. Sci. USA 2016, 113, 3603–3608.
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates G(D2) Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946.