2. Involvement of EWS-FLI1 in Transcription, Epigenetic Reprogramming, and Alternative Splicing in EwS
2.1. EWS-FLI1 in Transcription and Epigenetic Reprogramming
EWS-FLI1 is an aberrant transcription factor that drives cellular transformation by rewiring the epigenome to induce a large number of oncogenes. The N-terminus of EWSR1 contains a prion-like domain, characterized by an intrinsically disordered structure and low complexity. This domain has phase transition properties and manipulates multiple proteins involved in epigenome reprogramming and epigenetic alterations [
29,
30,
31,
32,
33,
34,
35,
36]. In addition to the canonical ETS-binding sites, EWS-FLI1 binds to DNA sequences at the GGAA/T core motif [
37,
38,
39] via a conserved ETS domain. It regulates multiple proteins through its prion-like domain to tumor-specific enhancers and promotors, recruiting acetyltransferases and establishing de novo enhancers by generating H3K27ac, thus opening the chromosomal architecture, which contributes to the activation of target genes [
29,
30,
37,
39]. The EWS-FLI1 protein complex includes RNA polymerase II [
23,
40], the core subunit hsRBP7 (human RNA polymerase II) [
41,
42], E2F3 [
43,
44], EWSR1 [
45], CBP/p300 [
46], WDR5, ASH2, MLL [
30], and the BAF complex (mammalian SWI/SNF complex) [
29,
47]. The threshold of GGAA motifs optimal for maximal expression is 20–26 [
48], which differs from that in wild-type FLI1. Super-enhancer-associated MEIS1 and RING1B also contribute to the chromatin reprogramming through co-localization with EWS-FLI1 at the active enhancers to drive the malignancy of EwS [
49,
50]. This specific coupling results in the activation of many genes (
Figure 1), such as NKX2.2 [
51], NROB1 [
52,
53,
54], IGF1R [
55], BCL11B [
56], EZH2 [
36], VRK1 [
30], GLI1 [
57], PTPL1 [
58], PPPR1A [
59], ERG2 [
60], GSTM4 [
61], PAX7 [
62], CHM1 [
63], REST [
64], PHF19 [
32], STEAP1 [
65,
66], SLFN11(Schlafen 11) [
67], HDAC3 [
68], TNC [
69], APCDD1 [
49], IL1RAP [
70,
71], MYC [
72], and PRC1 (protein regulator of cytokinesis 1) [
73].
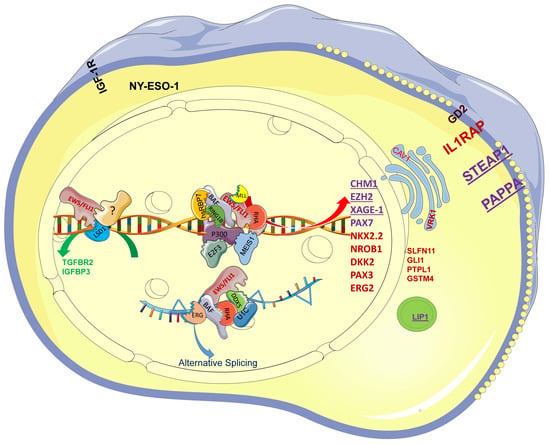
Figure 1. The EWS-FLI1 protein complex drives the specific transcription profile of EwS. EWS-FLI1 recruits E2F3, hsRBP7, BAF, RING1B, RHA, P300, and MEIS1, among others, to GGAA repeats and further activates CHM1, EZH2, PAX7, NKX2.2, NROB1, and STEAP1, among others. EWS-FLI1 functions as a protein complex with ERG, BAF, RHA, DDX5, and U1C to drive alternative splicing. Among the genes, the purple ones, such as CHM1, could serve as TCR-based immunotherapy targets; the red ones, such as NKX2-2, serve as diagnostic markers in clinic diagnosis. EWS/FLI1 recruits LSD1 and unknown transcription factors (?) to repress TGFBR1 and IGFBP3, which still needs further research.
Among the direct targets of EWS-FLI1, NKX2-2 mediates oncogenic transformation via transcriptional repression and is necessary and sufficient for the oncogenic phenotype of EwS [
74]. Further work demonstrates that NKX2-2, KLF15, and TCF4 occupy similar super-enhancers and promoters, forming an inter-connected auto-regulatory loop and occupying 77.2% of promoters and 55.6% of enhancers shared with EWS-FLI1 [
75], such as STEAP1 [
76]; this kind of coordinate regulation drives the proliferation of EwS. NROB1 directly interacts with EWS-FLI1 to modulate multiple gene expressions and mediate the oncogenic phenotype of EwS [
77]. SLFN11 is a putative DNA/RNA helicase that recruits to the stressed replication fork and irreversibly triggers replication block and cell death. Overexpression of SLFN11 is associated with resistance to topoisomerase I inhibitors and poly (ADP-ribose) polymerase (PARP) inhibitor combinations [
67,
78]. STEAP1 and IL1RAP are vital for the redox homeostasis of EwS [
65,
70]. APCDD1, PHF19, GSTM4, and PTPL1 are genes that are involved in the proliferation of EwS.
EWS-FLI1 is also involved in transcriptional repression of tumor suppressors such as IGFBP3 [
79] and PHLDA1 [
53] to drive oncogenic transformation [
31,
80]. The nucleosome remodeling and deacetylase (NuRD) complex is a typical ATP-dependent chromatin remodeling complex [
81] that plays a critical role in transcription and determines differentiation and development [
82]. EWS-FLI1 recruits the NuRD-LSD1 complex to repress LOX and TGFBR2 [
80,
83]. It also affects the transcriptional activation of AP-1 [
33] and MRTFB [
84] and binds to the promotor of FOXO1 to repress its expression, thereby increasing tumor growth [
85]. EWS-FLI1 promotes the phosphorylation of cyclin-dependent kinase-2 and AKT to inhibit the activity of FOXO1, thus rewiring transcriptional repression [
85]. EWS-FLI1 is also involved in the regulation of microRNAs (miRNAs) [
86]. It downregulates miRNA-145 to initiate mesenchymal stem-cell reprogramming toward EwS stem cells [
87] and represses miR-708, which induces the overexpression of EYA3 and contributes to the chemoresistance to etoposide and doxorubicin [
88].
The histone methyltransferase EZH2 exhibits silencing activity via methylation of H3K27 [
89]. In EwS, EWS-FLI1 upregulates EZH2 expression by interacting with the EZH2 promoter, thereby promoting tumor growth/metastasis and blocking endothelial/neuro-ectodermal differentiation [
36].
MiR-34a inhibits the proliferation and increases the sensitivity of EwS to doxorubicin and vincristine and is a strong predictor of a favorable prognosis in EwS [
90]. However, the exact mechanism underlying its downregulation remains elusive. Exportin 5 (XPO5), which mediates the nuclear export of pre-miRNAs and short hairpin RNAs [
91,
92,
93], interacts with EWS-FLI1 based on mass spectrometry [
94]. XPO5 is highly expressed in various cancers including EwS. Furthermore, the phosphorylation of XPO5 alters the nucleus and cytoplasm shift [
95]. Investigating XPO5 and its relationship with EWS-FLI1 may offer new insights into the therapy of EwS. Post-translational modifications of EWS-FLI1 modulate its transcriptional activity. Phosphorylation and O-GlcNAcylation of the N-terminus of EWSR1 [
96,
97,
98], as well as acetylation of the C-terminal FLI1 domain by PCAF (KAT2B, lysine acetyltransferase 2B), enhance the transcriptional activity of EWS-FLI1 [
99]. However, PCAF expression is lower in EwS tissues, which is a common feature of cancer.
2.2. EWS-FLI1 in Alternative Splicing
Pre-mRNA splicing is critical for gene expression, and most protein-encoding transcripts are alternatively spliced to provide diverse functions [
100,
101]. The N-terminus of EWSR1 interacts with the hyperphosphorylated RNA polymerase II and recruits serine-arginine (SR) through its C-terminus. After chromosome translocation, the C-terminus of wild-type EWSR1 is replaced by FLI1, which hinders the recruitment of SR-splicing factors and interferes with mRNA splicing [
23], thus demonstrating the negative property of this chimeric protein [
102]. This leads to comprehensive alternative splicing of numerous genes. Meanwhile, EWS-FLI1 interacts with the splicing components (snRNP) U1C and SF1 to modulate pre-mRNA splicing [
103]. It also recruits the BAF complex to drive the alternative splicing of ARID1A and the preferential splicing of ARID1A-L, which is necessary for tumor growth [
104]. Work by Selvanathan [
94] demonstrates that EWS-FLI1 is involved in the alternative splicing of CLK1, CASP3, PPFIBP1, and TERT, which potentially regulate the oncogenesis of EwS.
3. The Regulation of EWS-FLI1
Transcription and post-transcriptional modifications are involved in the regulation of expression and activity of EWS-FLI1. Although the transcriptional regulation of EWS-FLI1 remains elusive, the BRD4 inhibitor JQ1 suppresses this activity [
32,
34,
105]. HDAC6 deacetylates specificity protein 1 (SP1), thereby inhibiting the recruitment of the SP1/P300 complex to the promoters of EWSR1 and EWS-FLI1 and downregulating EWS-FLI1 [
106]. MiR-145 and let-7 repress EWS-FLI1 by targeting its mRNA [
87,
107,
108,
109] and inhibit the proliferation of EwS. The RNA-binding protein LIN28B interacts with EWS-FLI1 transcripts to maintain the stability and ensure the expression of EWS-FLI1 to enhance the tumorigenicity of the self-renewal of EwS [
108]. At the post-transcriptional level, EWS-FLI1 degradation is proteasome dependent, and the protein has a half-life of 2–4 h [
110]. This process can be protected by the action of ubiquitin-specific protease 19 (USP19) at the N-terminus [
111] and accelerated by tripartite-motif-containing 8 (TRIM8) at K334 [
112]; however, USP19 is expressed at low levels in EwS. Casein kinase 1 (CK1)-mediated phosphorylation of the VTSSS degron in the FLI1 domain activates speckle-type POZ protein (SPOP) activity, which degrades EWS-FLI1. In contrast, OTU-domain-containing protein 7A (OTUD7A) participates in the deubiquitination of the C-terminus and stabilizes EWS-FLI1 [
113]. The inhibitor of chromosomal maintenance 1 (CRM1 and XPO1), KPT-330 [
114], and IFN-γ [
115] suppress expression of EWS-FLI1 at the protein level. FOXM1, a downstream factor of EWS-FLI1, upregulates its expression [
116]. Cytosine arabinoside (ARA-C) downregulates EWS-FLI1 at the protein level and inhibits tumor growth [
117]; however, it shows hematologic toxicity and minimal activity in patients [
118].
STAG2 (stromal antigen 2) is a core subunit of the cohesion complex and is frequently mutated in multiple cancers [
119] including EwS [
10,
120]. Mutation of STAG2 in EwS is associated with poor outcomes by improving metastasis [
10]. Mechanically, in addition to the disruption of PRC2-mediated regulation of gene expression in EwS [
121], the inactivation of STAG2 strongly altered CTCF-anchored loop extrusion and decreases promotor-enhancer interactions. As a result, the cis-mediated EWS-FLI1 activity at GGAA microsatellite neo-enhancers is downregulated and the cells are enhanced in their migration and invasion properties [
121,
122].
Unlike STAG2, there was no evidence showing mutations of the ETS transcription factor ETV6 in EwS [
123]. ETV6 does not change the expression of EWS-FLI1 but co-occupy loci genome wide at the short consecutive GGAA repeats and constrains the transcriptional activity of EWS-FLI1 [
124,
125]. Upon inactivating ETV6, EWS-FLI1 overtakes and activates these cis-elements to promote mesenchymal differentiation by upregulating the expression of SOX11 [
125].
4. CAR-T Therapy
Unlike TCR-based T-cell therapy, which is limited to specific HLA restriction and deficient HLA expression in EwS [
205] because of the presence of myeloid-derived suppressor cells, F2 fibrocytes, and M2-like macrophages in the microenvironment [
190], CAR-engineered T-cell therapy can target specific cell-surface antigens in tumors, independent of HLA. VEGFR2 is a potential target for CAR-T-cell therapy in EwS [
206]. In addition to TCR-T-cells, CAR-T-cells [
207] can target STEAP1, which is involved in the malignant phenotype of EwS [
65].
CAR-T targeting GPR64, ROR1, and IGF1R, which are highly expressed in EwS [
137,
208], leads to a selective killing of EwS in vivo [
209]. LINGO1, which is highly expressed in EwS [
210], is a direct target of EWS-FLI1 (
Supplementary Figure S4B). EZH2 inhibition by GSK-126 induces GD2 surface expression in EwS [
211], and the combination of CAR-T therapy targeting GD2 and EZH2 inhibitors have synthetic cytotoxic in the treatment of EwS; this kind of combination provides new options for the clinical application. IL1RAP, a direct target of EWS-FLI1, is highly expressed in EwS, but minimally expressed in normal tissues, which makes it a promising surface target for EwS [
70] and a potential candidate for advanced CAR-T therapy. ICAM-1 can promote tumor cell/T-cell interaction and T-cell activation, and the knockdown of EWS-FLI1 upregulates ICAM-1 expression and leads to the upregulation of PD-L1 and PD-L2, both proteins that inhibit the activity of T-cells [
115]. Blocking PD-1 with a checkpoint inhibitor could increase the T-cell-mediated killing of EwS cells with low expression of EWS-FLI1.