Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Poly(ADP-ribose) (PAR) Polymerase 1 (PARP-1), also known as ADP-ribosyl transferase with diphtheria toxin homology 1 (ARTD-1), is a critical player in DNA damage repair, during which it catalyzes the ADP ribosylation of self and target enzymes. While the nuclear localization of PARP-1 has been well established, recent studies also suggest its mitochondrial localization.
- Poly(ADP-ribose) Polymerase 1
- PARP-1
- DNA repair
1. An Enzymatic Overview of Poly(ADP-ribose) Polymerase 1
The human PARP superfamily of enzymes currently has 17 known members. PARPs are involved in a variety of cellular functions, but are most famous for their involvement in DNA repair [14]. The name “PARP” is inaccurate for most PARP family members as only four enzymes catalyze ADP-ribose into a polymeric chain [15], while twelve are mono-ADP-ribosyl transferases (MARTs), and one has no catalytic activity. The key feature that allows these 17 proteins to be grouped into a single family is a conserved ADP-ribosyl transferase (ART) fold, which is located in a C-terminal domain in the 16 members, except for PARP-4, which is near the N-terminus [14,16].
The DNA damage response PARPs, PARPs 1–3, are the most well-known. PARP-1 generates the majority of intracellular PARylation (~90%) [17]. PARP-1 and PARP-2 play important roles in embryonic development and a PARP-1/PARP-2 double knockout is lethal, causing death at gastrulation [18]. Contrarily, PARP-1/PARP-3 double knockout is not lethal in murine cells [19]. All three of the DNA damage response PARPs have a high degree of similarity at the C-terminus, and the major difference occurs at the N-terminus: PARP-1 contains three N-terminal Zinc finger domains (Zn1, Zn2, and Zn3) and a domain containing a BRCT fold (BRCT), while PARP-2 and PARP-3 have a singular N-terminal domain (NTD) [20]. The C-terminal domains of the three proteins are highly similar, containing a WGR domain (rich in Trp, Gly, and Arg residues) and a catalytic (CAT) domain comprised of a helical subdomain (HD) and an ART fold (Figure 1A). The mode of DNA binding differs between PARP-1 and PARP-2; while the three major domains (NTD, WGR, CAT) work collaboratively to bind DNA in PARP-2, they contribute little to the DNA binding affinity in PARP-1, and its primary binding energy is contributed by Zn1 and Zn2 (Figure 1B), which are absent in PARP-2. Individual PARP-2 domains showed lower binding affinity than the full-length (FL) protein, regardless of DNA construct, as in [21]. Contrarily, the PARP-1 Zn1-Zn2 fragment displays an affinity for DNA that is comparable to FL, while the C-terminal region (Zn3, BRCT, WGR, and CAT) was unable to bind DNA effectively [22]. The DNA-binding mechanism of PARP-3 has not been explored to the same extent as PARP-1 and PARP-2, but current studies seem to indicate PARP-3 would recognize DNA in a manner similar to PARP-2. For both PARP-2 and PARP-3, their N-terminal regions are not strictly required for DNA-dependent activation, and the WGR domain is essential for catalytic activation in the presence of DNA [20].
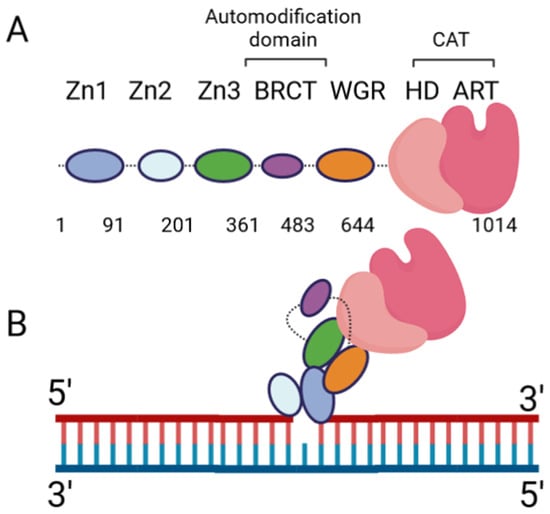
Figure 1. PARP-1 domain layout and arrangement in the DNA-bound form. (A) Cartoon representation of the six PARP-1 domains, connected by flexible linkers (dotted lines). (B) Cartoon representation of PARP-1:DNA complex, using a single-nucleotide-gapped DNA. Zn2 binds the 3′ end of the gap, while Zn1 binds the 5′ end. Zn1 serves as a scaffold for the assembly of the remaining domains in the appropriate orientation for catalytic activation.
Extensive biochemical and structural studies have answered key questions regarding PARP-1′s catalytic activation. First, it was established that PARP-1 required DNA binding to become catalytically active. It was later determined that four of the six domains are strictly required for catalytic activation: Zn1, Zn3, WGR, and CAT [23]. Zn1 makes extensive contacts with the DNA backbone and forms interfaces with the other three essential domains (Figure 1B). Ultimately, an internal auto-inhibitory helical subdomain (HD), which resides within the larger CAT domain, destabilizes, allowing sufficient space for NAD+ binding to the active site [23]. Hydrogen–deuterium exchange mass spectrometry (HXMS) experiments showed that, following DNA binding, the HD undergoes local unfolding, specifically in two of the helices (αB and part of αF), which otherwise prevent the active site from readily binding NAD+ [24]. Zn2 and BRCT were shown to be disposable for catalytic activation, somewhat ironically, since BRCT is a major site of automodification [25]. Nevertheless, in the absence of BRCT, the enzyme is still able to catalyze auto-PARylation. Solution NMR studies have shown that, while not necessary for activation, Zn2 plays an important role in DNA binding [26]. While DNA binding is critical for robust PARP-1 activation, PARP-1 binds to DNA in a sequence-independent manner [22]. PARP-1 binds to a wide variety of DNA structures, including blunt ends, 3′ and 5′ overhangs, with or without 3′ or 5′ modifications, and gapped DNA [20], as well as non-B form DNA such as DNA hairpins, cruciform DNA, and stably unpaired regions of DNA [27]. PARP-1 does show a preference for damaged DNA, preferentially binding a nicked DNA over circular, undamaged DNA [28]. While the kinetics and overall catalytic activity are affected by the DNA structure, PARP-1 activation can be triggered simply by the presence of exposed nucleotides and/or distortions in the helical backbone [27]. This provides a rationale for the involvement of PARP-1 in a wide variety of DNA repair processes [29].
During catalysis, PARP-1 chemically cleaves the nicotinamide moiety from nicotinamide adenine dinucleotide (NAD+). To avoid confusion, the two ribose groups as “adenosine-proximal” and “adenosine-distal” were designated based on their location relative to the adenosine group, which remains present through the reaction (Figure 2, left). Within the PARP-1 active site, three chemically distinct reactions occur, all of which covalently link the adenosine-distal ribose of the NAD+ in the active site: (1) initiation, linkage to an amino acid receptor; (2) elongation, linkage to the adenosine-proximal ribose of the nascent ADP-ribose chain; (3) branching, linkage to the adenosine-distal ribose of the nascent ADP-ribose chain [30]. The rate-limiting step in this process is initiation, while elongation is >200-fold faster and processive [31,32].
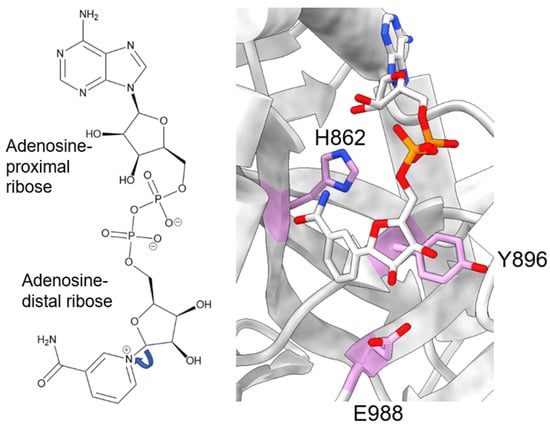
Figure 2. NAD+ molecule and its position in the PARP-1 active site. Left: Chemical structure of NAD+, with the two ribose moieties labeled as described in the text. Right: Zoomed-in view of a NAD+-analogue (benzamide adenine dinucleotide) bound to the PARP-1 active site (PDB: 6BHV). The H-Y-E triad of PARP-1 is shown in plum.
Initiation primarily involves the HYE catalytic triad, consisting of H862, Y896, and E988 [33,34,35]. H862 is involved in substrate binding and positioning, forming a hydrogen bond with the adenosine ribose (Figure 2, right), and mutants show significantly lower affinity for NAD+ (H862A) [34] or a lack of detectable activity (H862D) [36]. Y896 plays a role in substrate binding by stacking with the nicotinamide moiety (Figure 2, right) [35], though no published articles have examined mutations to this residue. E988 is implicated in substrate positioning, forming a hydrogen bond with the nicotinamide ribose (Figure 2, right), and mutations E988Q and E988K inactivate the elongation activity of PARP-1 [30,33,34]. During initiation, the acceptor amino acid must be nucleophilic. Indeed, D and E residues have long been considered the primary acceptors of ADP-ribosylation, owing to both their chemical nature and mass spectrometry-based detection methods [37,38]. However, other amino acids, such as K, R, S, T, C, and Y, have been shown to be modified [15,38,39,40,41,42], suggesting PARP-1 has an underlying mechanism for the enzymatic deprotonation of amino acids, which are non-nucleophilic under physiological conditions. Many ADP-ribosyl transferases capable of modifying arginine residues, which are also not nucleophilic at physiological pH, contain a catalytic dyad of E-X-E [43], while PARP-1 only has a single E residue in this pocket. An accessory protein, histone PARylation factor 1 (HPF1), was shown to switch the substrate specificity from D/E to S residues [44] by inserting a catalytic glutamic acid residue, HPF1 E284, into the PARP-1 active site, allowing this to work with PARP-1 E988 to allow the efficient catalysis of Serine ADP-ribosylation [45,46]. Still, in mass spectrometry studies using highly purified recombinant PARP-1, without adding HPF1, serine, lysine, and arginine, ADP-ribosylation was observed [38,41]. As these residues are not nucleophilic at physiological pH, the question of how these studies were able to observe the modification of non-nucleophilic residues becomes pertinent. E988 is in the appropriate position to act as a catalytic base, yet PARP-1 E988 mutants retain a minor amount of catalytic activity [30,33,34]. At the time of those studies, the ability of PARP-1 to modify serine residues was not as prominent of an idea, and the ability of E988 mutants to retain some level of catalysis was thought to reflect the nucleophilic nature of the D/E residues. PARP-1 E988 mutants, E988Q, E988K, E988A, and E988D, maintained the ability to perform initiation, albeit less robustly [30,34]. Kinetic assays interrogating E988A, E988Q, and E988D showed some impact, albeit mild, of the E988 mutations on the rate of initiation, with WT > E988Q > E988D > E988A [34]. It is interesting to note that the length of the amino acid correlates well with the initiation rate, rather than the charge of the amino acid. Based on the differences in initiation rates for the different mutations, it is possible E988 plays a non-essential role in initiation. The mechanism by which PARP-1 catalyzes ADP-ribosylation on non-D/E residues, in the absence of partner proteins, remains elusive.
Elongation involves H862, Y896, and E988 again during the catalytic step. M890 and Y986 are also involved during elongation, helping stabilize the adenosine and the pyrophosphate moieties, respectively, on the previous ADP-ribose unit [35,47,48]. In this step, E988 is crucial. E988D forms PAR less robustly and at a slower rate. E988A showed the minor elongation of MAR into oligo-ADP-ribose (OAR) at the longest time point assessed. E988Q did not show elongation activity [34], nor did E988K in a separate study [30]. Thus, E988 is playing a more active role in this enzymatic process. Mutations to Y986 impacted elongation and branching, with average PAR chains sizes of ~11 for Y986S, ~15 for Y986H, and ~38 for wild-type [30]. Interestingly, Y986S had the same amount of branching detected, while Y986H showed a 15-fold increase in branching [30]. The mechanism for the involvement of Y986 in branching is unknown. The elongation and branching involve the same glycoside bond, but occur on different riboses on the nascent chain. This minor difference could be related to the acceptor ADP-ribose binding in the wrong (opposite) orientation.
Whether linear or branched, the resulting enzymatic product is a PAR chain, which can impact enzymes in a covalent or non-covalent way. Non-covalently, PAR is a signaling molecule that recruits enzymes to sites of DNA damage, impacting their subcellular localization and allowing the efficient repair of single- and double-stranded breaks [29,49,50,51]. Essentially, enzymes involved in genome maintenance can have an intrinsic affinity for PAR, which can come through a PAR-binding motif (e.g., histones, p53, XRCC1, Lig3, DNA polymerase ε) [52], a macro domain (e.g., macroH2A, PARP-9, PARP-13) [53] containing a WWE domain (PARP-12, E3 ubiquitin-protein ligase (RNF146)) [54], or via a PAR-binding zinc finger domain (aprataxin polynucleotide kinase like factor (APLF), checkpoint with forkhead and ring finger domains (CHFR)) [55,56]. Alternatively, PAR can be covalently linked to target enzymes, which affects enzyme electrostatics, as an average PAR chain is ~38 ADP-ribose units in length [30], and there are two negative charges per ADP-ribose. Proteomics experiments have identified many protein targets for intracellular PARylation, primarily involved in chromosome organization, transcription, DNA repair, and mRNA processing [37]. Thus, PARylation can impact protein behavior by manipulating electrostatics and/or subcellular localization.
PARylation can be reversed by poly(ADP-ribose) glycohydrolase (PARG) [57] or another ADP-ribosyl hydrolase (such as ARH3, etc.) [53,56]. These enzymes can partially or fully remove the PAR chains from the modified molecule, and the balancing of PAR polymerase and PAR (glyco)hydrolase activities is required for many cellular processes, namely, DNA repair.
2. The Debate Regarding Mitochondrial PARP-1
PARP-1 is a prominent DNA repair enzyme in the nucleus, where it helps improve the efficiency of single-strand break repair and plays a role in other DNA damage response mechanisms [50]. The highly oxidative environment of the mitochondria leads to high levels of DNA damage, which must be quickly repaired, but how the organelle is able to efficiently clear DNA damage is unclear [5,6]. Some groups have explored whether PARP-1 plays a role in DNA damage repair in mitochondria. PARP-1 lacks a canonical mitochondrial localization sequence, so it had been unclear how PARP-1 could be imported into mitochondria. However, ADP-ribosylation has been observed in mitochondria, and much controversy has arisen regarding whether PARP-1 is the enzyme responsible for this observation.
Mitochondrial ADP-ribosylation was first described in rat liver mitochondria and the enzyme system reportedly generated protein-linked oligomeric ADP-ribose, though the enzyme class was not elucidated (e.g., NAD+ glycohydrolase vs. ADP-ribosyl transferase) [58]. Further studies would corroborate the finding of ADP-ribose in mitochondria, localizing it to the inner mitochondrial membrane, but the enzyme class was still unclear [59]. A mitochondrial ADP-ribosyl transferase was determined to be responsible for the protein ADP-ribosylation rather than the glycohydrolase, as reactions performed following the removal of the glycohydrolase still produced oligo-ADP-ribosylated proteins [60]. Similarly, ADP-ribosyl transferase activity was found in synaptic and nonsynaptic mitochondria purified from rat brain [61]. Building upon this work, a ~110 kDa protein capable of auto-ADP-ribosylation was described, but the authors were dissuaded from publishing these data, allegedly due to the general disbelief in mitochondrial ADP-ribosylation [62]. Later, the immunostaining of Sertoli and HeLa cells would show PARP-1 present in mitochondria at significantly higher concentrations than the surrounding cytoplasm [63]. The lack of any canonical mitochondrial localization sequence could explain the skepticism regarding mitochondrial PARP-1. Indeed, the question of how PARP-1 would enter the mitochondria was outstanding for many years. In 2009, Mitofilin, a transmembrane protein in the inner mitochondrial membrane, was identified to be responsible for transporting PARP-1 into mitochondria using immunoprecipitation–mass spectrometry, confocal laser microscopy and Western blotting methods; the depletion of Mitofilin abrogated PARP-1 mitochondrial import [64]. Other groups were able to find PARP-1 in mitochondria using proximity ligation assays, showing that PARP-1 interacted with both Exonuclease G (ExoG) and Pol γ in mitochondria [65]. Disease states, such as Chagas disease, or genotoxic stresses, like hydrogen peroxide treatment, can affect the concentration of PARP-1 in mitochondria [66]. The synthesis of a mitochondria-targeting PARP-1 inhibitor, XJB-Veliparib, inhibiting PAR formation in the mitochondria clearly illustrates the presence of PAR in mitochondria [67]. Liquid chromatography–mass spectrometry showed XJB-Veliparib specifically in mitochondrial fractions. Therefore, XJB-Veliparib, a specific PARP-1/2 inhibitor, was transported into the mitochondria, where it impaired intramitochondrial PARylation. The role of PARP-1 in mitochondria is being explored, and evidence supports a role for PARP-1 in mtDNA maintenance [64,65,66] and transcription [68].
On the other side of this argument, in a study investigating PARP-1-induced cytotoxicity in mouse embryonic fibroblasts treated with the DNA damaging agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), the authors performed subcellular fractionation and Western blots, and only found PARP-1 in the nuclear fraction, but not the mitochondrial in both control and MNNG-treated cells [69]. Despite not finding PARP-1 in the mitochondrial fraction, the study clearly illustrated that PARP-1 activation led to apoptosis-inducing factor (AIF) being released from mitochondria, and therefore the nuclear PARP-1 was said to be impacting the mitochondria. In another study using HeLa cells to study the cellular impacts of PARP-1 hyperactivation following exposure to MNNG, the authors utilized similar experiments, also failing to find PARP-1 in mitochondria. This study added to the field that PARP-1 and PAR were exclusively nuclear, seeing no signal for either in the cytosol or mitochondria [70]. Although the authors only found PARP-1 in the nucleus, they noted a decrease in mitochondrial ATP concentration following PARP-1 activation, again suggesting that the activation of nuclear PARP-1 impacts the mitochondria indirectly. A study exploring the localization of PARG in mouse cortical neurons, using the same major experiments for determining subcellular localization, only saw PARP-1 in the nucleus as well [71]. Similar experiments were performed on HeLaS3 and HEK293 cells [72], and also saw no PARP-1 in mitochondria or cytosol, but did observe oligo-ADP-ribose in mitochondria. More recently, it was shown that mitochondrial PARylation did not decrease in U2OS cells treated with inhibitors specific for PARP-1/2, and that U2OS PARP-1 knockout cells showed an increase in mitochondrial PARylation relative to the control [73]. In this study, the presence of PAR in mitochondria is established, contradicting the above studies. It was not stated that the subcellular localization for the PARP inhibitors was evaluated, and whether those inhibitors would be taken into the inner mitochondrial matrix is unclear. While all of these studies show no PARP-1 in mitochondria, they imply an intricate crosstalk between the nucleus and the mitochondria, whereby nuclear PARP-1 activation has an impact on mitochondrial homeostasis. Some of these studies utilized MNNG to damage the DNA, but whether this is transported into the inner mitochondrial matrix and damages the mtDNA at a rate comparable to nuclear DNA is not discussed. Additionally, these studies do not agree on the degree of mitochondrial ADP-ribosylation. Studies using DNA-damaging agents that specifically damage mitochondria DNA would be highly informative for the argument against PARP-1 in mitochondria.
Currently, the field is divided regarding whether PARP-1 localizes to mitochondria. Many studies utilize imaging-based techniques, such as the generation of fusion proteins followed by fluorescence microscopy or immunofluorescence. Experiments in which cells are transfected with vectors expressing exogenous fusion proteins are blind to the subcellular localization of the endogenous protein, and can result in artifacts caused by protein overexpression. In immunofluorescence experiments, artifacts can arise based on antibody quality, epitope availability in the cell (e.g., if a binding partner covers up the epitope), and the fixation method used. A systematic study comparing the subcellular distribution of proteins using both immunofluorescence and fusion proteins found that 82% of proteins tested showed “similar” localizations, with “similar” being defined as “one localization observed with both methods but with additional localizations in either of the two methods” [74]. For this reason, some authors decided to avoid imaging-based techniques and analyze PARP-1 subcellular localization using highly purified mitochondria [66,68]. Even in these cases, when Western blots are used to determine subcellular localization, artifacts can occur due to antibody quality and titer.
The debate regarding whether PARP-1 localizes to the mitochondria continues, as evidenced by the number of the above articles that were written within three years of this one. Some studies positively identify PARP-1 in mitochondria, while others do not see this enzyme localized to the mitochondria organelle under their experimental conditions. The identification of a transmembrane protein that imports PARP-1 into the mitochondria, Mitofilin, and the data suggesting that PARP-1 impacts the repair and transcription of mtDNA provide convincing evidence for PARP-1 in mitochondria. These data are bolstered by the in situ assays showing an interaction between PARP-1 and mitochondria-specific proteins within the mitochondrial matrix. The mechanisms governing the PARP-1/Mitofilin interaction remain unexplored, and would provide insights into the conditions required for PARP-1 import into mitochondria. The studies in which PARP-1 was not seen in mitochondrial fractions could suggest that PARP-1 import requires specific cell types and/or specific conditions, as opposed to being constitutively found in mitochondria.
This entry is adapted from the peer-reviewed paper 10.3390/biom13081195
This entry is offline, you can click here to edit this entry!