Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
|
Pathology
There has been a boost in autophagy reports due to its role in cancer progression and its association with tumor resistance to treatment. The availability of large cancer datasets has provided an extensive evidence-based approach to understanding the role of autophagy-related genes in various human cancers and their clinical implications, including cancer progression, development, and treatment response.
- autophagy
- cancer
- gene
- Data analysis
1. Introduction
To assess the clustering of solid tumors, researchers utilized the expression levels of autophagy-related genes. To ensure robust observations, researchers focused on solid tumor types with more than 100 tumor and control samples. Employing this approach, researchers generated a UMAP plot that revealed three distinct clusters among the sixteen solid tumors analyzed (Figure 1A). The identified clusters were as follows: Cluster 0 comprised BRCA, KIRC, KIRP, LGG, KIHC, LUAD, LUSC, and PRAD; Cluster 1 included COAD, ESCA, PAAD, and STAD; and Cluster 2 consisted of GBM, OV, SKCM, and TGCT. Notably, cluster 0 grouped tissues with similar genetic or anatomical profiles, such as BRCA-PRAD, KIRC-KIRP, or LUAD-LUSC. Cluster 1 predominantly encompassed gastrointestinal tumors, while Cluster 2 included TGCT and OV, which are tumors from reproductive organs. To identify differentially expressed genes characterizing these clusters, researchers identified 18 genes that primarily distinguished Clusters 0 and 1. Cluster 2 exhibited decreased levels of these markers (Figure 1B), and therefore it will not be analyzed in further detail herein. Nevertheless, it is important to note that autophagy profiles have been induced and studied on SKCM, GBM, and OV models with anti-tumoral effects [29,30,31,32]. In addition, for GBM and SKCM, there is possible to suggest that expression similarities in non-pivotal genes could be originated at their division from the ectoderm, as was demonstrated for the P2X7 receptor [33]. Then, these findings could support the evolutive hypothesis of cancer as an embryological phenomenon [34].
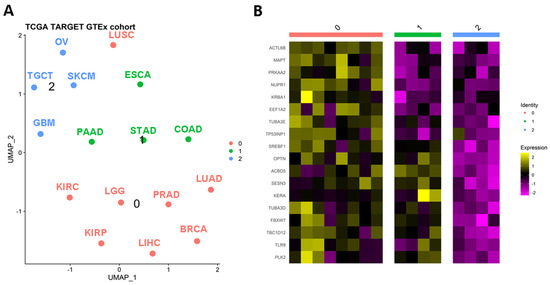
Figure 1. Autophagy-related genes can stratify solid tumors. (A) Clusterization of solid tumors based on the differential expression of autophagy genes. After a UMAP analysis, it is possible to recognize three classifications (B) of relevant tumors based on the expression of autophagy genes. BRCA: breast invasive carcinoma; COAD: colon adenocarcinoma; ESCA: esophageal carcinoma; GBM: glioblastoma multiforme; KIRC: kidney renal clear cell carcinoma; KIRP: kidney renal papillary cell carcinoma; LGG: brain lower grade glioma; LIHC: liver hepatocellular carcinoma; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; OV: ovarian serous cystadenocarcinoma; PAAD: pancreatic adenocarcinoma; PRAD: prostate adenocarcinoma; SKCM: skin cutaneous melanoma; STAD: stomach adenocarcinoma; TGCT: testicular germ cell tumors.
2. Autophagy Regulators Specific to Cluster 0
Researchers' analysis revealed that the genes ACTL6B, MAPT, PRKAA2, NUPR1, KRBA1, EEF1A2, TUBA3E, and TP53INP1 specifically characterized cluster “0” through their overexpression. Furthermore, researchers observed that these genes exhibited upregulated levels in tumors belonging to Cluster “0” compared to their normal adjacent tissues. While the limited number of normal-adjacent samples in the TCGA data introduces potential biases and limitations, the majority of the putative markers for Cluster “0” could be validated using the UALCAN tool [35].
2.1. Protein and Mutational Features of Relevant Genes for Cluster 0
To gain insight into the protein products of these genes, researchers utilized the UALCAN tool to examine their change in proteins with data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) cohort. In the LIHC dataset, researchers found a comparable group of normal-adjacent samples, which demonstrated the upregulation of EEF1A2, NUPR1, and MAPT proteins in the tumor group. However, there were some inconsistencies. For instance, while the PRKAA2 gene exhibited notable overexpression in LIHC, its corresponding protein was significantly downregulated. This disparity suggests the importance of considering the mutational profile of these genes or post-translational events on the produced proteins. Based on data from cBioPortal [36] of the TCGA datasets belonging to Cluster “0”, these genes exhibited a low frequency of mutations (1.1–5%), implying that somatic mutations may not play a significant role in dysregulating the relationship between autophagy-related genes and their corresponding proteins.
2.2. Previous Research on Relevant Genes for Cluster 0
According to the MSigDB, the NUPR1 and PRKAA2 genes participate in macroautophagy and its regulation, while MAPT serves as an autophagy regulator, TP53INP1 contributes to autophagosome organization, and ACTL6B and KRBA1 are involved in the autophagy-related network. Additionally, other genes are associated with less-studied forms of autophagy. For example, EEF1A2 is linked to chaperone-mediated autophagy and its regulation, PRKAA2 is involved in lipophagy-related pathways, and TUBA3E is associated with aggrephagy.
Previous studies have highlighted the significant role of NUPR1 in macroautophagy and its impact on the aggressiveness and treatment resistance of specific tumors such as BRCA, LUAD, LUSC, LIHC, and LGG [37,38,39,40,41,42,43]. NUPR1, also known as p8, is a transcriptional regulator that has been shown to reduce apoptosis caused by dihydroartemisinin (DHA), sorafenib, or ionizing radiation (IR) in LIHC tumor cells [37,38,39]. However, opposing effects have been observed in osteosarcoma and non-tumor cells [44]. Additionally, research has demonstrated that Δ9-tetrahydrocannabinol (THC) induces autophagy-mediated apoptosis in an LGG model [40]. Despite autophagy-related pathways being upregulated in LIHC and LGG tumors, it remains uncertain whether these pathways promote tumor growth or tumor suppression, necessitating further investigation. In lung and breast cancers (LUAD, LUSC, and BRCA), repression of NUPR1, in combination with conventional anticancer therapies, has been proven to control tumor growth [41,42,43]. Another study supports the inhibition of NUPR1 using microRNA-637 (hsa-miR-637) as a promising option for this purpose [45].
Controversial results have emerged regarding the expression of the PRKAA2 gene (Protein Kinase AMP-Activated Catalytic Subunit α 2, AMPKα2) in gastrointestinal malignancies [46,47,48,49]. Some studies suggest that repression of PRKAA2 promotes tumor growth in gastrointestinal cancer by suppressing ferroptosis, an autophagy-dependent form of cell death [46]. On the other hand, other studies propose that PRKAA2 activates autophagy-related pathways, leading to treatment resistance, and that its activation can be triggered by the gastrin hormone [47,48,49]. In LIHC, inhibiting PRKAA2 has been shown to downregulate autophagy rates, and metformin has been identified as a potential PRKAA agonist for controlling hepatitis C virus (HCV) replication [50]. In glioma, low expression of PRKAA2 has been associated with a better prognosis [51]. Although these findings may seem contradictory, especially considering that LGG belongs to Cluster “0”, they underscore the importance of including autophagy-related factors in the intra- and inter-individual heterogeneity of tumors.
The MAPT gene encodes the microtubule-associated protein tau, which has been extensively studied in Alzheimer’s disease (AD) [52,53]. Recent research has explored the interplay between autophagy and MAPT in AD and has demonstrated that overexpression of MAPT/tau inhibits the fusion of autophagosomes with lysosomes, leading to autophagosome accumulation through increased levels of LC3 protein [54]. Although direct links between MAPT and autophagy in cancer remain limited, the high expression levels of Tau protein in glioblastoma, a tumor with enhanced autophagy activity, have raised questions about its possible role in oncogenesis and its implications for cancer therapy [55].
In BRCA cohorts, a long non-coding RNA (lncRNA) for the MAPT gene called MAPT-AS1 has been found to be overexpressed in tumor tissues [56,57,58]. This is noteworthy because lncRNAs, which are usually located in antisense strands of DNA from original genes, can also be affected by somatic mutations irrespective of their canonical effects. Thus, the combination of somatic mutations and non-coding RNA as potential prognostic markers deserves further attention, as demonstrated in COAD [59].
TP53INP1 gene exhibits inconsistent findings across experiments and tumor tissues. Some researchers have identified hsa-miR-106a as an oncomiR that targets TP53INP1 in metastatic lung cancer [60], indicating its involvement in tumor suppression. Increasing the levels of TP53INP1 could be crucial in controlling tumor growth through autophagy-dependent cell death. In the case of PRAD, hsa-miR-30a and hsa-miR-205 have been suggested as potential therapeutic options for suppressing TP53INP1 [61,62]. However, it has been explained that TP53INP1 is overexpressed as a response to ionizing radiation, which confers resistance [61,62]. Therefore, suppressing this gene could potentially resensitize tumor cells to standard treatment protocols. Like other representative genes in this cluster, TP53INP1 exhibits a dual function. According to Peuget et al. (2021), oxidative stress induces the expression of TP53INP1 [63]. This stress can trigger autophagy by interacting with LC3 in the cytoplasm or apoptosis by interacting with P53 in the nucleus, and the role of mitochondria and their metabolism in this process is also implicated [64]. Thus, an additional factor to consider in researchers' analysis is the localization of autophagy-related transcripts and proteins. Unfortunately, there is insufficient information available to conduct this type of comparison.
In summary, NUPR1, PRKAA2, TP53INP1, ACTL6B, KRBA1, EEF1A2, and MAPT genes are coexpressed with 17 other genes (ANK2, ST8SIA1, GUCY2F, HERC1, TRHR, COL11A1, CHRM3, CNR2, KITLG, ROR1, CDKL5, PPOX, IGF2R, DDIT3, OPCML, ELOVL5, and BRINP2) according to the GeneMania database [65]. These genes are enriched in the MAPK pathway (p = 0.004) [66], allowing us to associate cluster “0” with a MAPK-dependent macroautophagy-like process. However, it is important to note the significant heterogeneity observed in the samples, classifications, tumor tissues, and other forms of autophagy.
3. Tumors Balancing Macro- and Micro-Autophagy Processes (Clusters 0 and 1)
Clusters “0” and “1” in Figure 1 represent a distinct group of genes associated with tumors that exhibit a balance between macroautophagy and microautophagy processes. Notably, the genes SREBF1, OPTN, ACBD5, SESN3, KERA, TUBA3D, FBXW7, TBC1D12, TLR9, and PLK2 show high expression levels in various tumors such as ESCA, PAAD, STAD, COAD, LUAD, LUSC, KIRC, LGG, PRAD, KIRP, LIHC, and BRCA.
In addition, after performing a random forest Gini importance analysis, researchers observed that KERA, TP53INP1, SREBF1, and TUBA3E showed great accuracy (above 75%) and over 75% of Gini contribution. It suggests the potential contribution of these autophagy-related genes in the classification of tissues regarding their dysregulation between tumor and normal samples.
Of particular interest are the TUBA3D and FBXW7 genes, which are associated with the chaperone-mediated protein folding pathway (R-HSA-390466) according to the Enrichr database [66]. This suggests their potential involvement in chaperone-mediated autophagy. Supporting this idea, these genes have also been implicated in certain forms of microautophagy, such as aggrephagy and mitophagy, as indicated by the MSigDB. Additionally, these genes are part of the regulatory pathways of macroautophagy along with the other eight genes that cluster these tumors. ACBD5, SREBF1, and OPTN genes are also involved in microautophagy pathways, including aggrephagy, mitophagy, and xenophagy.
3.1. Accumulation of ACBD5 Is Found in Tumors from Cluster 0 and 1
Notably, the ACBD5 gene is interesting in autophagy-related studies as its deregulation can induce their accumulation at protein levels. This gene has been associated with peroxisome maintenance, lipid exchange, and homeostasis, which are crucial processes for lipid and carbohydrate metabolism reorganization in tumor cells [67,68]. These processes involve microautophagy pathways such as pexophagy, aggrephagy, and mitophagy [69].
3.2. Previous Research on Overexpressed Genes in Tumors of Clusters 0 and 1
Other genes related to microautophagy processes include PLK2, SESN3, TLR9, OPTN, and SREBF1. Independent research has demonstrated that the PLK2 gene controls α-Synuclein aggregation in an autophagy-dependent context [70]. Although this process is dependent on macroautophagy and regulated by mTORC1 inhibition, it appears to be a microautophagy pathway that is specifically activated in the presence of its substrate, α-Synuclein [70,71]. An interesting regulatory axis involves the lncRNA OIP5-AS1, which targets hsa-miR-126 to prevent α-Synuclein aggregation in autophagy-activated cells [71].
Regarding the SESN3 gene, recent studies have identified its role as an autophagy activator in tumor cells by repressing mTORC1 [72]. However, this gene has also been associated with other autophagy pathways such as chaperone-mediated autophagy [73]. Overexpression of SESN3 has been observed in LUAD [73] and ESCA [74] models, suggesting its potential involvement in promoting pro-tumor autophagy pathways. Expression levels of this gene can be regulated by specific miRNAs, such as hsa-miR-194-3p [73] or hsa-miR-429 [74].
About mitophagy, several reports have described the upregulation of the TLR9 gene in tumors belonging to Clusters “0” and “1” [75,76,77], indicating its involvement in inducing this form of autophagy. In BRCA, it has been reported that this gene plays a role in the rewiring of doxorubicin and may explain the cardiomyocyte death and systolic dysfunction observed in patients undergoing this tumor treatment [78]. Consistent with these findings, TLR9 was found to be upregulated in aggressive versions of LIHC, LUAD, LUSC, and COAD models [79,80,81]. Consequently, various regulatory pathways have been proposed to control TLR9 expression. For example, hsa-miR-30a has been shown to sensitize LIHC cells to a combined therapy of hydroxychloroquine and sorafenib by repressing TLR9 [79]. On the other hand, inducing TLR9 expression in dendritic cells has been suggested as a potential therapeutic strategy, as demonstrated in PAAD cases [82]. It is important to note that bulk analyses using next-generation sequencing (NGS) do not differentiate between the origins of cells within tumors, which can lead to different interpretations of the results. Therefore, researchers are increasingly turning to single-cell sequencing to differentiate immune cells, tumor cells, and normal-adjacent cells with varying autophagy-related profiles within the same tumor pool.
In addition to TLR9, OPTN has been extensively studied in the context of mitophagy. PINK1 and PRKN, which are highly studied autophagy-related genes, are also involved in this process. The PHB2 gene stabilizes PINK1 in mitochondria, facilitating the recruitment of Parkin (the product of PRKN), ubiquitin, and optineurin (the product of OPTN) to promote mitophagy [83,84,85,86]. However, a recent study challenges the necessity of PINK1 and PRKN for initiating mitophagy [87]. Consequently, it has been suggested that OPTN may have tumor suppressor functions by activating suppressor autophagy mediated by HACE1, a tumor suppressor [88,89,90], or by repressing the pro-oncogenic transforming growth factor-β (TGFβ) signaling in triple-negative breast cancer (TNBC) cells, a subtype of BRCA [91]. Importantly, OPTN has been found to be downregulated in GBM tumor samples, which has been corroborated by independent studies [92]. The same study proposes that inducing OPTN expression in GBM cells could help control tumor growth, supporting a suppressive role for this gene, although the underlying mechanism remains unknown.
In terms of the application of OPTN in the context of mitophagy and the tumor environment, several studies have identified OPTN as a potential therapeutic target. For instance, it has been observed that OPTN induces pro-tumor mitochondrial-related autophagy, reducing the efficacy of combined treatments involving pemetrexed, cisplatin, and MEK inhibitors or anti-PD-L1 in a LUSC model [80]. In a PAAD model, repression of OPTN leads to apoptosis through chaperone-mediated autophagy [93]. Similar to TLR9, understanding the function of OPTN allows us to differentiate its contribution to tumor growth based on its expression in surrounding cells. In LUAD models, higher expression of OPTN in fibroblasts surrounding the tumor contributes to tumor invasiveness [94].
SREBF1 upregulation has been linked to mTORC1-dependent autophagy, which may be induced by leptins to suppress ferroptosis in BRCA, LIHC, PRAD, and LUAD models [95,96,97,98]. Additionally, SREBF1 levels were found to be elevated in PAAD tissues, regulated by high glucose concentrations. In PAAD models, the upregulation of SREBF1 helps control autophagy levels [99]. This gene may act as a negative regulator of mTORC1-dependent autophagy, favoring pro-tumor microautophagy pathways. It is worth noting that SREBF1 can function as both a protein and a transcription factor. Studies have demonstrated that genes upregulated by the SREBF1 transcription factor can be altered in the presence of cisplatin, inducing treatment resistance in a LUSC model [100]. This evidence highlights the importance of carefully analyzing autophagy-related genes with dual functions to enhance researchers' understanding of this process. A study proposed that mTORC2 stabilizes SREBF1 through FBXW7-mediated regulation to integrate autophagy and lipid metabolism processes, leading to the downregulation of target genes such as acetyl-CoA carboxylase and fatty-acid synthase [101].
Considering the combined findings of two genes involved in tumor clusterization, FBXW7 and SREBF1, it is hypothesized that these tumors exchange autophagy-related processes and large-scale technologies based on their aggressiveness and treatment sensitivity or resistance. However, conducting large-scale high-throughput analyses in mass groups could obscure specific autophagy pathways in certain tumor subtypes or patients. Therefore, the current perspective is to compare global observations with focused research. Nevertheless, the scientific community is moving towards a comprehensive analysis of tumors, considering their heterogeneity and subclonal profile, which will allow us to confirm researchers' current hypotheses about autophagy-related processes in the tumor environment in the future.
Regarding macroautophagy, the FBXW7 gene has been the focus of numerous studies aiming to characterize its function. This gene is known as a tumor suppressor as it is frequently mutated or suppressed in human tumors [102]. However, its dysregulation in chemoresistance remains controversial, suggesting that its behavior depends on the context. It has been observed to be upregulated in resistant gastric cells [103] and downregulated in chemoresistant models of BRCA [104].
Interestingly, FBXW7 has been found to induce the expression of ATG16L1, an important gene involved in LC3 lipidation and autophagosome formation, while not affecting the levels of other autophagy-related genes (ATG) [105]. Moreover, FBXW7 suppresses mTORC1, thereby activating autophagy pathways [106,107]. FBXW7 participates in different molecular axes, resulting in different effects on tumor cells. For instance, the GSK3-FBXW7 interaction leads to the ubiquitination and degradation of Rictor, increasing cellular ROS (reactive oxygen species) in an autophagy-activated context [108]. On the other hand, interactions between FBXW7 and oncogenes such as SHOC2 or LSD1 can reduce the expression of autophagy-related pathways [106,107,109]. In conditions where tumors are growing, cisplatin treatments have been shown to induce the degradation of the MRE11-RAD50-NBS1 (MRN) complex by FBXW7 and lysosomes [102]. As a result, the overexpression of the MRN complex or the suppression of the FBXW7 gene can lead to cisplatin-resistant tumors and a poor prognosis. In relation to this, hsa-miR-25 and hsa-miR-223 have been shown to suppress FBXW7 levels, promoting autophagy and treatment resistance in LIHC [110] and LUAD [111] models, respectively. Anti-miRs could be used to counteract the suppression of FBXW7 levels, but it is important to better understand the specific context in which this strategy would be applicable.
Lastly, three genes (TBC1D12, KERA, and TUBA3D) that contribute to tumor clustering in groups “0” and “1” have not been previously associated with the tumor-related autophagy process. It is important to emphasize that, in researchers' analysis, the KERA gene was the top gene in Gini relevance and accuracy in tissue pooling of groups between 0 + 1 vs. 2. Studies on mutations in the TBC1D12 gene (TBC1 Domain Family Member 12) have been conducted in urological tumors, suggesting that alterations in its mutational profile could be linked to worse patient survival [112]. Interestingly, this gene exhibits a higher mutation frequency in PRAD samples compared to other patients. The KERA (Keratan Sulfate Proteoglycan Keratocan) gene has been found to have lower levels in cisplatin and paclitaxel-resistant OV models [113], partially aligning with observations in the entire dataset (Cluster “2”). The expression levels of the TUBA3D (Tubulin α-3D Chain) gene in BRCA (upregulated) and OV (downregulated) have been validated [114,115]. Notably, in BRCA models, TUBA3D was shown to be downregulated in paclitaxel-resistant cells compared to parental cells [116].
In summary, the findings presented in this discussion suggest that all the aforementioned genes may make significant contributions to tumor-related autophagy through their expression in tumors and the surrounding cells, warranting further attention in future research.
This entry is adapted from the peer-reviewed paper 10.3390/genes14081550
This entry is offline, you can click here to edit this entry!