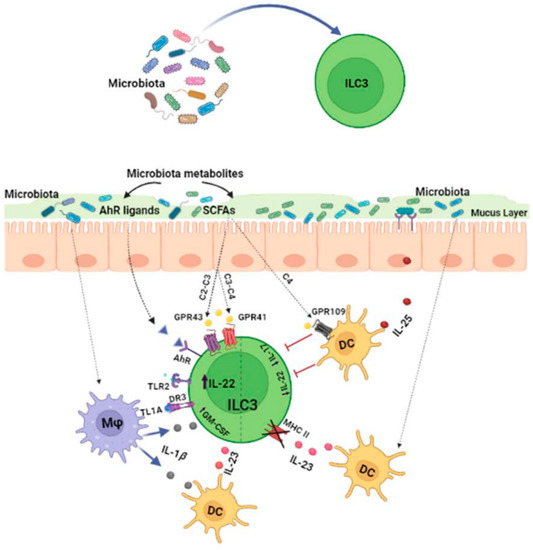
1.1. Effects of the Microbiota on ILC3s
Commensal microbes have a crucial impact on ILC3 behavior, both through direct stimulation and, indirectly, the regulation of epithelial and myeloid cell responses [
13]. Signals derived from commensal microbes can be directly sensed by specific immune cell receptors, such as toll-like receptors (TLRs). Spits and co-workers detected TLR transcript encoding for types 1, 2, 5, 6, 7 and 9 (but not 3 or 4) in human ILC3s [
14]. However, they reported that only TLR2 was functional and that its stimulation could induce IL-2 that acted in an autocrine manner to increase IL-22 expression by ILC3s [
14]. Besides TLRs, RORγt
+ ILCs can directly recognize commensal bacteria through the engagement of natural cytotoxicity receptors (NCRs), such as NKp44, NKp46 and NKp30, which have been recently identified as another pathway for the direct recognition of PAMPs (pathogen-associated molecular patterns) [
15,
16]. Among NCRs, NKp44 triggering induces TNFα release by ILC3s, whereas the combined engagement of NKp44 with cytokine stimulations are able to induce IL-22 secretion [
17,
18].
Gut microbiota-derived tryptophan metabolites, such as indole and indole-3-acid-acetic (IAA), are ligands for the aryl hydrocarbon receptor (AhR), a ligand-dependent transcription factor expressed by RORγt+ ILCs. AhR activation is essential for the development and function of intestinal RORγt+ group 3 ILCs [
19]. AhR activation promotes IL-22 expression through cooperative interactions with RORγt, which directly binds at the IL-22 promoter to induce its transcription [
20]. AhR-mediated IL-22 production by ILC3s promotes protective responses following DNA damage in epithelial stem cells and the postnatal development of isolated lymphoid follicles and cryptopatches. In another study [
21], an AhR agonist, in combination with IL-1β, stimulated ILC3s to produce IL-22, whereas an AhR antagonist caused the differentiation of ILC3s into IFNγ-expressing ex-ILC3s, confirming the conserved function of AhR in supporting ILC3 maintenance. Additionally, AhR activation supports the expansion of ILC3s promoting the transcription of Notch 1/2 and the expression of anti-apoptotic proteins, such as Bcl-2 and Ki67 [
22]. In Ahr
−/− mice, both the percentage and absolute number of RORγt
+ ILCs are significantly decreased, indicating that AhR deficiency could lead to the defective expansion of ILC3s [
19]. Metabolites generated by the fermentation of dietary components by the microbiota could be ligands for specific receptors expressed on the surface of ILC3s. Among these, short-chain fatty acids (SCFAs), such as acetate (C2), propionate (C3) and butyrate (C4), are known to influence ILC3 functions since they express G protein-coupled receptors (GPCRs). ILC3s are able to sense SCFAs via Ffar2 (GPR43), Ffar3 (GPR41) and GPR109a [
23]. Ffar2 mainly regulates CCR6+ ILC3 function, which is a highly proliferative ILC3 subset in colonic cryptopatches, isolated lymphoid follicles and larger colonic patches [
24]. In particular, acetate mainly promotes ILC3 proliferation via Ffar2 through the activation of the AKT, STAT3 and mTOR signaling pathways, whereas propionate preferentially stimulates IL-22 production via Ffar3. IL-22 production is also indirectly supported by acetate via the upregulation of IL-1β receptor expression on ILC3s. In line with these findings, it has been reported that the genetic ablation of Ffar2 (Ffar2
ΔRorc) significantly impairs the proliferation and production of IL-22 by CCR6
+ ILC3s, resulting in hindered host defense and reduced tissue repair [
24]. Butyrate promotes IL-22 release by RORγt
+ ILCs through the GPR41 pathway [
25] and can inhibit IL-17-producing ILC3s by interacting with GPR109a, which is expressed by dendritic cells (DCs) [
26]. In addition, microbiota-derived butyrate is a regionally specialized factor suppressing ILC3s in terminal ileal Peyer’s patches (PPs). Indeed, GPR109a is expressed highly in RORγt+ PP ILC3s and butyrate treatment significantly reduces the amount of IL-22 produced by ILC3 PP cells [
27]. Additionally, GPR109a signaling negatively regulates ILC3 expansion. In mice treated with niacin, a Gpr109a agonist, the frequency of ILC3s is reduced, whereas the deletion of this receptor is associated with higher numbers of ILC3s [
25]. Another report showed that the
Helicobacter Species functions as a negative regulator of ILC3s in the large intestine. In particular,
H. typhlonius and
H. apodemus promote the loss of colonic T-bet- and RORγt-expressing ILC3s, thereby inducing gut dysbiosis [
28].
The microbiota can also influence ILC3 activity through the modulation of myeloid and epithelial cell responses in the GI tract [
29]. Indeed, the microbiota triggers IL-1β production through the Myd88- and Nod2-dependent pathways via intestinal CX3CR1+ macrophages. This cytokine can induce the production of GM-CSF from ILC3s or IL-23 via CD103+ CD11b+ DCs, which increases IL-22 production by ILC3s [
30]. Upon microbial stimulation, CX3CR1+ macrophages can also release the TNF-like ligand 1A (TL1A), whose binding with death receptor 3 (DR3) expressed on human ILC3s promotes not only their proliferation but also their IL-22 production when combined with cytokine stimulation [
31,
32]. Overall, the microbiota is able to sustain IL-22 production by ILC3s through various mechanisms; however, on the other hand, the same microbiota tightly regulates this cytokine by constantly limiting its release to prevent the deleterious effect of IL-22. Mechanistically, the microbiota can induce IL-25 release by epithelial cells, which reduces the amount of IL-22 produced by ILC3s in a DC-dependent manner. The inhibitory mechanism mediated by IL-17RB+ (IL-25R) on DCs requires physical interactions with ILC3s and is independent from IL-23 [
5].
Interestingly, commensal bacteria can also directly affect the antigen presenting capability of ILC3s. In particular, under steady-state conditions, the microbiota can induce IL-23 production by mononuclear phagocytes (DCs and macrophages), a crucial signal for the reversible silencing of MHCII+ in ILC3s, thereby reducing the capacity of ILC3s to present antigens to T cells in the intestinal mucosa [
33].
Thus, the gut microbiota can significantly affect ILC3 functions through various mechanisms (Figure 1), but further studies are still required to more specifically understand the microbial signals that activate, suppress or fine-tune ILC3s in the gut.
Figure 1. The microbiota regulates ILC3 functions through various mechanisms. The microbiota and its metabolites can directly affect ILC3 activity through a diverse array of receptors expressed on their surfaces. Alternatively, microbes can modulate ILC3 functions by stimulating the release of cytokines by myeloid or epithelial cells.
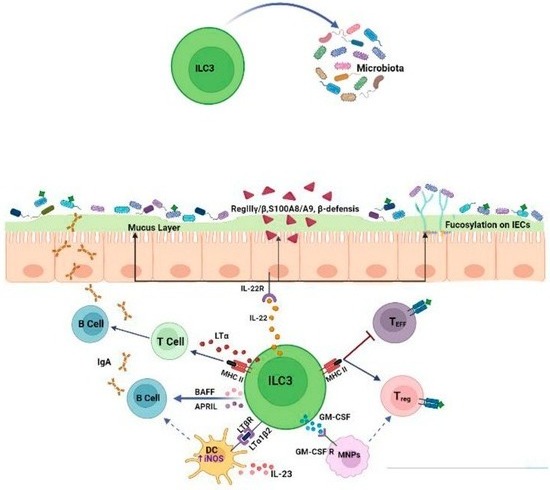
1.2. Effects of ILC3s on the Microbiota
ILC3s play a pivotal role in preserving the homeostasis of the gut microbiota by regulating physical, biochemical and immunologic barriers. In the intestinal tract, ILC3s are a dominant source of IL-22 and interact with intestinal epithelial cells (IECs) to modulate responses to the microbiota and promote bacteria anatomical containment [
34]. In this regard, Sonnenberg et al. [
11] demonstrated that Rag1
−/− mice that were treated with anti-IL-22 mAb showed the dissemination of
Alcaligens (commensal bacteria) from the intestinal lymph nodes to the spleen and liver, causing systemic inflammation and a loss of intestinal homeostasis. Another study [
35] showed that in IL 22
−/− mice, tight-junction proteins (claudine and occludine), mucins and antimicrobial peptides (S100A8 and S100A9, β-defensin, RegIIIβ, RegIIIγ) were reduced in epithelial cells, thus resulting in the loss of the physical separation of the microbiota from the intestinal epithelium [
36]. Moreover, IL-22 produced by ILC3s promotes intestinal epithelial fucosylation by inducing the expression of fucosyltransferase 2 (Fut2). This process offers survival advantages to beneficial bacteria that use fucose for their colonization and as an energy source. Indeed, in RORγt IL-22-deficient mice, Fut2 expression is significantly reduced and the mice become more susceptible to infection [
37]. Besides IL-22, ILC3s can also induce epithelial fucosylation via lymphotoxin α (LTα) production and, accordingly, the numbers of F-ECs and Fut2 expression are reduced in Lta
−/− mice. A positive feedback loop exists between membrane-bound LTβ (LTα1β2) and IL-22 since LTβ produced by ILC3s triggers IL-23 secretion by DCs, in turn inducing IL-22 production by ILC3s [
38]. So far, only a few studies have investigated gut microbial species in deficient mice or altered ILC3 populations. However, higher levels of
Segmented filamentous bacteria (SFB), as well as
Clostridiales species, have been reported in the absence of ILC3s (RORcCre × Id2fl/fl) [
21]. IL-22 and LTα produced by ILC3s can also influence microbiota composition [
37,
39]. In the absence of Ahr, outgrowths of SFB have been observed due to the loss of IL-22 produced by ILC3s, indicating the role of this lymphocyte population in the regulation of microbiota composition. Along the same line, in both Lta
−/− and IL-22
−/− mice, the expansion of SFB and a concomitant decrease in
Bacteroides have been found [
40], whereas in another study, IL-22 depletion resulted in a marked decrease in commensal
Lactobacilli [
35].
ILC3s can also interact with adaptive immune cells to modulate the quality and magnitude of immune responses to commensal microbiota. Recent studies have highlighted the critical role of ILC3s in negatively selecting microbiota-specific CD4
+ T cells to maintain intestinal tolerance. In particular, ILC3s can act as antigen-presenting cells by expressing high levels of MHC class II within mesenteric lymph nodes. MHCII has been found to be highly expressed on RORγt+ ILCs that lack the expression of both T-bet and NKp46 while exhibiting the homogenous expression of CCR6 and the heterogeneous expression of CD4 [
41]. However, different from classical antigen presentation, the interaction between CD4
+ T cells and MHCII+ ILC3s results in the suppression of effector CD4
+ T cells rather than their proliferation [
41]. Indeed, intestinal ILC3s lack the expression of canonical co-stimulatory molecules (CD40, CD80, CD86) and, different from circulating ILC3s, remain absent even upon stimulation by IL-1β [
42]. Moreover, ILC3s express CD25 molecules (conferring a high affinity to IL-2R), by which they can compete with T lymphocytes to bind IL-2, which is essential for their survival. In line with these observations, mice lacking MHC-expressing ILC3s exhibit increased frequencies of effector CD4
+ T cells against commensal microbiota, leading to spontaneous intestinal inflammation [
43]. To maintain immune tolerance against microbiota, antigen-presenting ILC3s are also able to induce microbiota-specific T regulatory (Treg) cells [
44]. To ensure Treg differentiation, the migration of ILC3s to mesenteric lymph nodes via CCR7 is necessary, as well as their expression of integrin, aVß3 through which they promote the activation of latent TGF-β, which is required for Treg differentiation [
45]. Interestingly, ILC3s produce large amounts of IL-2 in the gut, which is known to support Treg survival [
46]. In addition, ILC3-derived GM-CSF promotes Treg cell differentiation by acting on mononuclear phagocytes [
29]. ILC3s lacking one of these factors fail to maintain tolerance and lead to the expansion of pro-inflammatory Th17 cells against commensal bacteria [
47]. ILC3s can also present antigens to T follicular helper (Tfh) cells by modulating B cell responses in germinal centers [
48]. In the intestinal tract, the majority of activated B cells differentiate into IgAs producing cells, which is critical for mediating host–microbiota mutualism. IgAs promote physical segregation and shape the composition of microbiota by regulating bacteria colonization [
49]. The dysregulation of ILC3s results in altered class-switched B cells and an increase in the high-affinity IgA coating of potential colitogenic bacteria. In addition, ILC3s regulate the production of IgA through the release of LTα and membrane-bound LTβ [
40]. LTα sustains T cell-dependent IgA induction by favoring T cell homing to the gut, whereas LTβ interacts with DCs, upregulating their expression of inducible nitric oxide synthase (iNOS), which is necessary for IgA induction [
40]. Finally, ILC3s can directly support IgA production by releasing B cell survival factors, such as the B cell-activating factor (BAFF) and A proliferation-inducing ligand (APRIL) [
50].
Collectively, these data demonstrate that ILC3s are critical regulators of mucosal immune responses to microbiota (Figure 2), thus act as checkpoints to maintain host–microbiota homeostasis.
Figure 2. ILC3s regulate immune responses to commensal microbiota. ILC3s ensure the anatomical containment of bacteria, shape microbiota composition and orchestrate various tolerance mechanisms by acting on epithelial cells and adaptive immune populations.
2. Influence of ILC3–Microbiota Interactions on the Outcomes of ICI Therapy
Growing evidence has reported that the microbiota plays a crucial role in modulating ICI efficacy [
68,
69]. It has been reported that a “favorable” gut microbiome composition could enhance therapeutic response to ICIs, whereas antibiotic-treated patients and germ-free mouse models have shown a compromised efficacy of ICI therapy [
68,
69,
70,
71,
72,
73].
In a mouse model of colon cancer, the antitumor effect of CTLA-4 blockade was enhanced by specific
Bacteroides species, which are associated with Th1-specific responses mediated by IL-12-producing intratumoral DCs [
74]. Similarly,
A. muciniphila restores PD1 blockade responsiveness by increasing the recruitment of IFN-γ+ CD4
+ T cells within tumor microenvironments [
70]. Along the same line, the antitumor efficacy of both anti-CTLA4 and anti-PD1 treatments is supported by a specific mixture of bacteria strains that are able to increase the frequency of tumor-infiltrating GrB+IFNγ+CD8
+ T cells [
68], which in turn suppress tumor growth and prolong progression-free survival in mice.
Recently, in a CRC mouse model, Goc et al. [
52] demonstrated that the disruption of the dialog between MHCII+ ILC3s and T cells results in a microbiota compositional shift that favors immunotherapy resistance. This microbiota compositional shift concerns an unbalanced ratio between
Bacteroidales and
Clostridiales, which is associated with positive and negative ICI outcomes, respectively. In this regard, it has been reported that a decrease in either the number or activity of ILC3s results in marked reductions in commensal
Bacteroides and
Lactobacilli [
40], strains that are positively associated with ICI responsiveness in CRC [
75]. Importantly, alterations in ILC3/T cell crosstalk and immunotherapy resistance have already been associated within the context of chronic intestinal inflammation, such as inflammatory bowel diseases (IBDs), suggesting that the impairment of ILC3s could act in the very early stages of CRC development. ILC3–T cell interactions occur within TLS [
52], which, in the context of ICI therapy, are associated with the regression of neoplastic lesions [
76], longer disease-free survival and overall survival [
77,
78]. The density of TLS in CRC correlates with the frequency of ILC3s and with their expression of TLS formation-related genes [
53]. Tumor-infiltrating ILC3s can support lymphoid neogenesis [
18], promote vasculature changes [
79,
80] and favor the recruitment of leukocytes within tumors [
18,
81], suggesting that ILC3s could sustain TLS formation and affect therapeutic responses to ICIs. Moreover, within tumors, ILC3s are able to produce CXCL10 and recruit both CD4
+ and CD8
+ T lymphocytes, thereby promoting antitumor immune responses and enhancing the efficacy of checkpoint inhibition [
82].
As a whole, microbiota manipulation represents an interesting therapeutic option for overcoming immunotherapy resistance. Fecal microbiota transplantation (FMT) from patients who respond to immunotherapy into non-responders or refractory patients restores ICI efficacy through Th1-dominant antitumor immune responses [
70-
84]. Analysis of the fecal bacterial content of melanoma patients who responded to anti-PD1 treatment [
73] revealed the relative abundance of
Bifidobacterium. Interestingly, it has been reported that
Bifidobacterium species can produce aromatic lactic acids, including indolelactic acid (ILA), which can engage AhR, highly expressed by ILC3s, to mediate their immune responses. Short-chain fatty acid (SCFA) administration or diet integration with probiotics [
85] or fiber as pectin [
86], which enhances SCFA-derived microbiota production, can be used as interventional measures to promote ICI efficacy. SCFA administration combined with PD-1 therapy is associated with IFN-γ response [
87], higher frequencies of CD4
+ T cells [
88] and CD8
+ T cells [
89] and higher progression-free survival [
90]. SCFAs can have a direct effect on ILC3s expansion and functions in a GPCR-dependent manner [
25,
91] and it has been reported that the administration of SCFA-ameliorated anti-PD1/PD-L1 treatment reduces IL-17A-producing ILC3s [
92]. Although immunological information is still limited in this model, including IL-22 production by ILC3s, these findings indicate the potential involvement of ILC3s in the mechanisms underlying the efficacy of SCFA–ICI combined therapy.
Although growing evidence has suggested that ILC3s may be critically involved in the outcome of ICI treatment influenced by the microbiota, further studies are required to clarify the role of ILC3s in the mechanisms underlying microbiota influence, aiming to improve both antitumor immune responses and immunotherapy efficacy in CRC.