Despite continued efforts, there remain no disease-modifying drugs approved by the United States Food and Drug Administration (FDA) or European Medicines Agency (EMA) to combat the global epidemic of Alzheimer’s disease. Currently approved medicines are unable to delay disease progression and are limited to symptomatic treatment. It is well established that the pathophysiology of this disease remains clinically silent for decades prior to symptomatic clinical decline. Identifying those at risk of disease progression could allow for effective treatment whilst the therapeutic window remains open for preservation of quality of life. This review aims to evaluate critically the current advances in the interpretation of tau-based biomarkers and their use to provide insights into the onset and progression of Alzheimer’s disease, whilst highlighting important future directions for the field
- Tau
- biomarkers
- diagnostics
1. Introduction
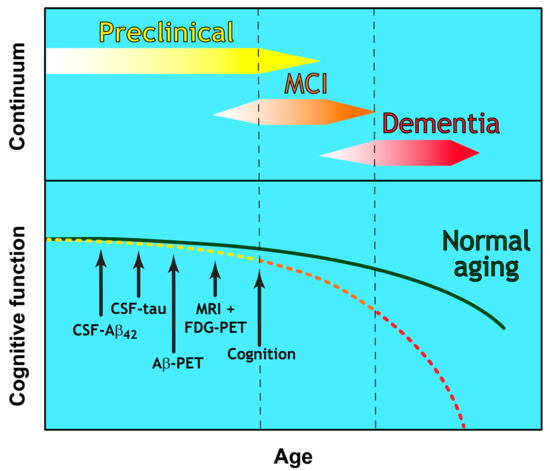
Until recently, a diagnosis of probable AD was based largely on clinical history and psychometric testing. These document memory impairment and deficits in a range of cognitive functions, with a requirement for disease pathology at post-mortem to confirm a definite diagnosis [7]. This early diagnostic framework was revised by the National Institute of Ageing-Alzheimer’s Association (NIA-AA) in 2011 with two notable updates. The first was the introduction of biomarker testing to support clinical diagnostic criteria by including analysis of cerebrospinal fluid (CSF) amyloid β and tau. The second was the recognition that the development of AD is a continuum that could be defined as three clear stages of disease (as described in [8] and Figure 1):
-
Preclinical Alzheimer’s disease [8]
-
A disease stage based entirely on biomarker-based changes
-
No clinical symptoms (presymptomatic)
-
Currently offers no diagnostic utility for clinicians
-
-
Mild Cognitive Impairment (MCI) due to Alzheimer’s disease [9]
-
Established criteria for the prodromal symptomatic phase
-
AD biomarker evidence may be used to support diagnosis
-
-
Dementia due to Alzheimer’s disease [10]
-
Refined clinical criteria for the demented phases of AD
-
Biomarker evidence may be used to support diagnosis
-
2. Diagnostic Utility of Tau within CSF
2.1. Total Tau and Truncated Tau
Total tau (T-tau) levels in the CSF can distinguish between healthy individuals, those with MCI and AD dementia patients. An increase in CSF T-tau is also predictive of future conversion to later disease stages [41]. Since there is a high degree of heterogeneity at the protein level between various tauopathies, and since CSF T-tau is elevated following neuronal injury caused by other factors such as traumatic brain injury [42] and acute stroke [43], it remains to be seen whether T-tau alone will be useful in a diagnostic setting [44]. Whilst T-tau is a validated clinical biomarker for the AD continuum, more research is required to unlock fully the true potential of tau as an AD-specific diagnostic biomarker. Currently most CSF T-tau tests, such as the AlzBio3 kit (Innogenetics), are based on detection antibodies around the N-terminal half of the tau molecule. These are unable to distinguish between full-length tau and fragments that lack N- or C-terminal regions [45]. This raises concern as to whether current T-tau tests are missing valuable information hidden within the molecule and whether these measurements are truly representative of “total tau”. One area of ongoing promising research is exploring the utility of tau fragments as biomarkers of the AD continuum.
There is growing evidence that truncated tau fragments are present in the AD brain (for an extensive review see [46]). There is a lack of consensus regarding the specific nature of these fragments and their role in disease pathology. Their identification and characterisation could greatly assist in the differential diagnoses of the tauopathies which are often difficult to distinguish at early disease stages. The core of the paired helical filaments (PHFs) which comprise neurofibrillary tangles is restricted to the repeat domain [47,48,49,50] implying that large portions of the C-terminal half of the molecule may be sequestered. Therefore, it may be important to consider the ratio of N- and C-terminal fragments as potentially indicative of the tangle burden within the brain. It has been shown that the majority of fragments within the CSF of AD patients are a mixture of smaller fragments with only a small proportion measurable by ELISA assays that depend on detection of full-length or even N-terminally intact tau [51]. This points to the need for continued research into the pathways that lead to the formation of these fragments in order to gain a better understanding of whether the generation of any specific fragments is upregulated and whether there are specific associations with disease onset and progression.
The majority of tau degradation and clearance is mediated by the proteasome and autophagic degradation systems [52]. It is likely that upstream post-translational modifications, aggregation and interactions with chaperone proteins govern how tau molecules finally arrive into these clearance systems. Numerous sites of truncation have been identified within the tau protein (Figure 2) but it is not yet understood how these arise or how they relate to the AD continuum. Many truncations arise as part of normal physiology but a better understanding of these degradative pathways in various conditions could give rise to the identification of truly disease specific tau fragments and disease specific AD biomarkers.
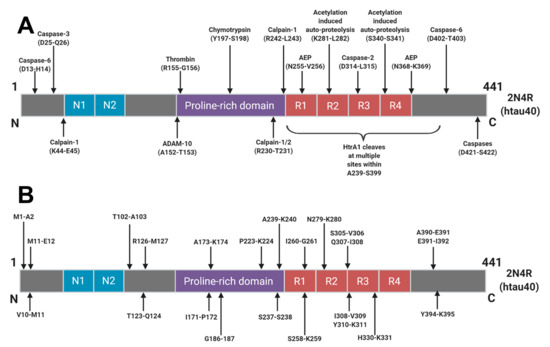
In CSF, several potential AD-specific fragments are being investigated and it is hoped that these will add to the refinement and diagnostic utility of T-tau measurements going forward. An N-terminally truncated fragment ending at Lys-224 was found to be specific to neurons, found in NFTs, and upregulated in AD [53]. Furthermore, a fragment ending at residue Asp-368, that is cleaved by asparagine endopeptidase (AEP), was found to be upregulated in AD CSF [54]. Only 3–8% of measurable tau in the sample had a C-terminus at residue Asp-368 but a shift to a higher proportion of this fragment in AD CSF would suggest that the fragmentation pattern may change in disease. Interestingly, levels of AEP were found to be lower in AD patients, which suggests that measuring the expression of various tau clearance mediators may have diagnostic merit. A separate study found the levels of a fragment ending at Gly-314, a C-terminally truncated soluble tau species that is responsible for cognitive impairment in transgenic mouse studies, to be higher in the brains of cognitively impaired individuals compared to normal counterparts [55]. The levels of this fragment were modestly predictive of overall cognitive impairment but interestingly the level of caspase-2, a mediator of proteolytic clearance, was found to be higher in cognitively impaired individuals as well. Several C-terminal fragments have also been found to be enriched in late stage disease [56]. Monomeric tau is preferentially cleaved by calpain-1 at a site within the core region of PHFs but upon oligomerisation, there is a shift in fragmentation pattern to a more C-terminally dominant conformation. These data suggest that a transient and malleable tau clearance mechanism might exist within the brain which may be readily altered in response to various stages of aggregation. It remains to be seen if these changes reflect tau clearance mechanisms which represent an adaptive response to pathology and how they contribute to the pathophysiology of the disease as it progresses in the brain.
From these selected publications, it is clear that numerous truncated fragments of tau exist and are linked to disease progression and severity. A greater understanding of this “tauosome” may provide an untapped wealth of information if we look beyond the current limited focus on T-tau measurements.
2.2. P-Tau
Phosphorylation plays an essential role in the physiological function of tau, predominantly by regulating axonal growth, plasticity, and transport through altering the microtubule binding affinity of tau [57,58,59]. There are approximately 85 potential phosphorylation sites spanning the full-length tau molecule, with as many as 49 phosphorylation sites detected in the AD brain (Figure 3).
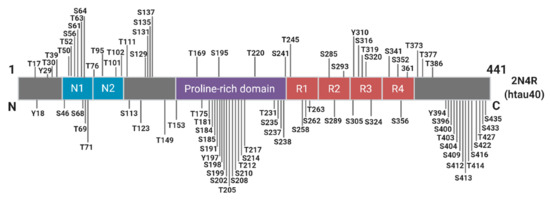
Figure 3. Schematic of Known Tau Phosphorylation Sites. Sites below the depicted 2N4R tau fragment have been identified in the Alzheimer’s brain. Information derived from [60].
Although the level of phosphorylated tau represents less than 5% of total PHF-tau [70] neocortical neurofibrillary pathology in AD correlates strongly with hyperphosphorylated tau load as determined by immunohistochemistry performed on post-mortem brain samples [71]. Importantly, phosphorylated tau load in the brain correlates with the concentration of phosphorylated tau in the CSF in vivo. It can therefore act as a reliable biomarker for AD and hence its inclusion as a diagnostic criterion [71].
2.3. Expanding from P-tau181
Despite the potential 85 phosphorylation sites spanning the tau molecule, most studies have focused solely on just one phospho-tau site, P-tau181. Other potential AD biomarkers such as P-tau231 [76] and P-tau199 [77] have been studied but not to the same degree as the “gold standard” P-tau181. Although P-tau181 offers clear evidence of diagnostic utility for AD, exploring other phospho-tau sites could further enhance this utility.
Several recent studies have also concluded that P-tau217 may offer improved diagnostic utility for the AD continuum especially when compared to P-tau181. P-tau217 was increased 6-fold in AD CSF when compared to non-AD samples, whereas this increase was only 1.3-fold for P-tau181. P-tau217 was also deemed a more specific marker than P-tau181 for the preclinical phase of AD as determined by its correlation with Aβ PET scans [80]. A similar study supports this data showing increases in CSF concentrations of P-tau217 in prodromal AD and AD dementia, which was several-fold higher than P-tau181 [81]. In addition, the authors also reported a greater longitudinal increase of CSF concentration and improved correlation with Aβ and tau PET imaging. A further related paper [82] quantified P-tau181, P-tau202, P-tau205 and P-tau217 signatures in CSF, across 35 years of the AD continuum. This study showed that P-tau217 and P-tau181 begin to increase around the same time as amyloid plaque formation is initiated, 21 and 19 years prior to symptom onset, respectively. At the time when neuronal dysfunction (measured using 18F-fluorodeoxyglucose-positron emission tomography [FDG-PET]) can be detected approximately 13 years prior to the appearance of clinical symptoms, P-tau205 begins to increase. Lastly, as neurodegeneration begins (as determined by clinical decline, initiation of the symptomatic phase and cortical atrophy shown by MRI), tau-PET ligands begin to detect filamentous tau deposits in tangles, with P-tau217 and P-tau181 gradually decreasing. It was also shown that P-tau202 does not increase throughout the course of the AD continuum [82].
To conclude, P-tau181 does not equate to P-tau and should not be reduced to this in the literature. Moving forward, P-tau could represent a component of a more comprehensive phospho-tau signature that encompasses several disease-specific phospho-sites that could help map the AD continuum through the preclinical, prodromal and symptomatic stages of the disease.
3. Translation of CSF AD Biomarkers to the Periphery
CSF biomarkers can give valuable information about the rate of protein production and clearance at a given point in time within the central nervous system (CNS). This has been shown to aid with the clinical diagnosis of other CNS disorders including multiple sclerosis [92], Guillain-Barré syndrome [93], cancers of the brain or spinal cord [94,95], and serious bacterial, fungal, and viral infections [96]. CSF biomarkers used for AD diagnosis have also proven to be a valid proxy for monitoring neuropathological changes in AD. CSF-based measurements involve potentially dangerous and certainly unpleasant lumbar punctures whilst PET imaging is limited to only the best
equipped medical facilities. Attempting to roll out a primary diagnostic screening framework for non-symptomatic and preclinical AD is simply not viable with these current tools. New approaches that allow rapid, low-tech analysis of biomarkers in more readily accessible fluids such as a fingerprick of blood, could be transformative for routine detection of AD at early disease stages. More invasive and hospital-based procedures could then be reserved for use as secondary and confirmatory diagnostic tools if needed. The holy grail of AD biomarker research therefore remains the provision of a cheap, accessible, straight-forward blood test that can reliably diagnose the AD continuum, or at the very least identify those who would require CSF or PET secondary testing.
3.1. From CSF to Plasma: T-tau, P-tau181, and Tau Fragments
The logical diagnostic journey for suitable blood-based AD biomarkers is to pursue the candidates that were validated first in CSF. The evidence for the use of T-tau as a plasma-based AD biomarker has been relatively robust to date, with a systematic review and metanalysis of 271 patients with AD (6 cohorts) and 394 controls (5 cohorts) showing average fold increase of 1.95 for T-tau over controls [72]. Since this analysis, plasma T-tau has been shown to be as strongly associated with AD dementia as CSF T-tau and higher plasma T-tau is associated with faster progression of AD [41,101]. However, the data are not conclusive with some studies showing no statistical difference or a large degree of overlap between control, MCI, and AD cohorts in terms of plasma T-tau levels [38,102]. Three independent studies have shown that P-tau181 in plasma can differentiate between AD, non-AD neurodegenerative disease and control subjects. Plasma P-tau181 was also shown to be increased in the earliest preclinical change of the AD continuum and correlated well with Aβ PET load [103–105]. An 8-year longitudinal arm of one study also showed that high plasma concentrations of P-tau181 were associated with cognitively unimpaired and MCI participants who went on to develop AD dementia [103]. A recent large study has shown high correlation between elevated plasma levels of P-tau217 and brain amyloid levels measured by PET [106]. By measuring plasma levels of P-tau217, this study differentiated AD patients from patients with other tauopathies and controls, with a significantly higher diagnostic accuracy than P-tau181, neurofilament light chain and MRI-based biomarkers. In addition, significantly higher levels of plasma P-tau217 were measured in PSEN1 mutation carriers compared to non-carriers. Participants in this cohort had a mean age of 35.8 and elevated P-tau217 levels were noted in individuals as young as 25 years of age, which amongst mutation carriers is 20 years prior to estimated onset of MCI. Of the few studies that have investigated the importance and diagnostic value of novel tau fragments in plasma, an N-terminal fragment ending at Ser198 was found to distinguish healthy controls from AD-MCI and AD patients [51]. This study could not distinguish between AD-MCI and AD cohorts suggesting the difference was linked to an early pathological event in the AD continuum that was soon saturated. Interestingly, this study also showed T-tau and N-terminal fragments ending at Lys224 did not differentiate between healthy controls, AD-MCI, and AD patients [51].
4. Conclusions
The ever-pressing challenge of diagnosing AD at the earliest possible stage of the disease process necessitates the pursuit and discovery of novel biomarkers. As we continue to validate novel biomarkers for AD, we expand our understanding of the molecular mechanisms of AD pathogenesis. This can translate into a more accurate diagnosis of disease stage, particularly in presymptomatic AD, and potentially permit monitoring of progression and response to treatment. The ultimate goal would be to define each stage of the AD continuum using a validated biological signature of ADspecific biomarkers. In doing so, AD treatment could permit a personalised and stratified therapeutic approach that is appropriate for each disease stage. Presymptomatic diagnosis may eventually allow early therapeutic interventions to interrupt the chronic neurodegenerative process of AD prior to any loss of life quality.
AD patient cohorts in these studies include patients with a clinical diagnosis of dementia, specified as sporadic or genetic AD. Early onset familial AD is caused by rare, dominantly inherited mutations in amyloid precursor protein (APP) and presenilin proteins (PSEN1 and PSEN2), and accounts for approximately 1% of all AD cases [117]. An interesting research avenue would be to compare the tau biomarker signature of patients with autosomal dominant familial AD to patients with sporadic AD.
Currently, only P-tau and T-tau are validated biomarkers for the AD continuum. The present authors believe that an individual’s “tauosome” signature, made up of a complex and diverse family of protein conformations, PTMs and fragments, could play an important role as a defining AD diagnostic biomarker. The translation of CNS-focussed tau biomarkers to the periphery is encouraging and could potentially pave the way for a more accessible primary screen to identify those at risk of becoming symptomatic.
The search for tau diagnostics in plasma is a rapidly expanding field with the potential to revolutionise the diagnosis of AD and other tauopathies. “Test, test, test” became the WHO and global mantra for overcoming the COVID-19 pandemic. A similar objective may contribute to beating back the relentless march of the already existing global AD epidemic. Removing the burden of this testing from specialist diagnostic units into a primary care setting may permit screening of far more people, giving us the best opportunity to preserve the healthspan of individual patients with disease modifying therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21228673