1. The Purpose of Metabolic Therapy in Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is a primary myocardial disorder characterized by enlarged ventricles with a contractile deficit that leads to reduced ventricular function in the absence of volume or to pressure overload, congenital heart disease (CHD), or significant coronary artery disease (CAD) [
1]. Genetic and non-genetic causes underlie its pathogenesis, in which a mixed mechanism is often involved [
2]. Its estimated prevalence in the general population ranges from 1:500 to 1:2500 [
3,
4].
DCM is a recognized cause of systolic heart failure (HF), a condition in which the impaired cardiac pump is unable to fulfil the energetic demand of the body in terms of nutrients and oxygen supply. HF has been defined as the cardiovascular epidemic of the 21st century [
5], with an estimated prevalence of about 2–4% in the general adult population; this prevalence is known to increase with age [
6,
7]. Moreover, due to the prevalence of HF in high-income countries, significant amounts of human and economic resources are employed in this field [
8].
Multiple factors, such as hemodynamics, neurohormones, and genetics, participate in progressive heart failure remodeling [
9]. The efficiency of the myocardial pump depends mainly on the metabolism of the cardiac cells [
10]. However, the hyperactivation of the adrenergic system and the renin–angiotensin–aldosterone system (RAAS), when attempting to sustain hemodynamic failure, contributes to indirect changes in the metabolism of the cardiac and skeletal muscles, worsening their efficiency [
11]. In the last decades, better knowledge of the pathophysiological mechanisms of the effects of the neurohormonal axis on the cardiovascular (CV) system has enabled the adoption of drugs that block this detrimental activation. Despite the development of additional molecules targeting different pathological pathways, as outlined by the European and American guidelines on HF [
12,
13], a specific phenotype-oriented therapy is generally lacking, and the HF prognosis remains poor [
14].
In the last decades, there has been a growing interest in the cardiac metabolism; the aim has been to find new potential disease biomarkers and therapeutic targets to improve the HF prognosis [
15].
A healthy adult heart produces adenosine triphosphate (ATP) by metabolizing different types of fuels (fatty acids, glucose, lactate, ketones, and amino acids), primarily via oxygen-dependent mitochondrial oxidative phosphorylation and the electron transport chain and, to a lesser extent, the anaerobic glycolytic pathway [
16]. Between 60 and 80% of the energy produced by the healthy heart is derived from free fatty acid (FFA) oxidation, despite a metabolic flexibility that allows a shift between different energy substrates to maintain ATP production [
16]. However, FFA oxidation is a less efficient source of energy production than glucose oxidation (in terms of the produced ATP per consumed O2 molecules). Of note, the amount of ATP produced per O2 molecules consumed is greater for glucose oxidation compared to that of FFAs. For example, the mitochondrial ATP yield per oxygen atom (P/O ratio) is only 2.33 for long-chain fatty acids, whereas it is 2.50 for ketone bodies and 2.58 for glucose. Consequently, oxidizing glucose results in an increase in cardiac efficiency of up to 30% [
17].
The failing heart is considered to be an “engine out of fuel” [
18]. A reduction in the mitochondrial oxidative capacity is the first metabolic change characterizing the heart’s deteriorating energy deficiency [
19,
20]. The ensuing increased glycolytic pathway [
21] is not as efficient as mitochondrial phosphorylation and fails to compensate for the status of the energy deficit; this also leads to H
+ accumulation in the cytoplasm as glycolysis is uncoupled from the oxidation of pyruvate and lactate [
22,
23]. Moreover, a highly significant shift in substrate utilization also occurs. A decrease in FFA oxidation has been observed in humans with idiopathic DCM [
24], despite evidence from other studies that is not consistent with this finding [
25,
26]. Furthermore, despite a reduction in FFA oxidation, the failing heart still mainly counts on this substrate for the highest proportion of mitochondrial ATP generation [
27].
Changes in substrate utilization in the failing heart have also been observed relative to glucose metabolism, ketone bodies, and long-chain amino acids [
15]. More metabolites have been found to be involved in the pathophysiological process of HF; thus, the studies on HF are no longer limited to glucose and FA metabolism [
28]. When glucose and lipid metabolism decrease, it is possible to hypothesize that ketone bodies may act as alternative substrates in failing hearts [
28]. Due to insulin resistance and other factors, the failing heart has significantly reduced glucose and fatty acid utilization, and ketone bodies are fast-metabolizing small molecular energy substrates that the heart can use to improve cardiac efficiency. In addition, recent research has demonstrated that branched-chain amino acids (BCAAs) also play a significant role in the pathophysiology of end-stage heart failure. The myocardial BCAA metabolism can effectively improve cardiac function and slow the progression of heart failure [
28]. Therefore, the metabolic remodeling of small molecular substrates such as ketone bodies and amino acids also plays a significant role in the onset and progression of HF, in addition to the changes in the metabolism of glucose and FAs during HF. However, there have been conflicting results; these results are partially explained by the disparities in the severity of the disease and the presence of other medical conditions, such as metabolic syndrome, in the individuals participating in human clinical studies.
In addition, in systolic heart failure, metabolic changes can occur regardless of weight status. However, the specific metabolic changes seen in obese, normal-weight, and underweight individuals may vary. There may be an increased reliance on fatty acid oxidation as a source of energy for the heart in obese people with heart failure. This can lead to an impaired glucose metabolism and decreased myocardial glucose uptake and oxidation. In addition, metabolic dysregulation in the heart may be exacerbated by insulin resistance and inflammation, which are frequently associated with obesity. As previously mentioned, heart failure can cause metabolic changes, such as an increase in glycolysis as a source of myocardial energy, even in healthy people. In heart failure, the oxidative metabolism is less efficient; so, this metabolic switch is made to make up for it. Heart failure patients who are underweight may experience metabolic changes that are comparable to those seen in people of normal weight. There may still be a shift toward more glycolysis [
29]. Implementing strategies for the targeted reduction in this particular fat store in obese individuals is the obvious solution to the problem of preventing adipose tissue inflammation and the metabolic and cardiovascular complications that come with it. The treatment for insulin resistance and obesity still relies heavily on lifestyle changes, such as diet and exercise modifications [
29].
Recently, the use of various “omics” technologies, such as metabolomics, has offered a new chance to enhance our understanding of the mechanisms involved in this disease and to find new biomarkers for the prognosis and diagnosis of DCM [
30]. A recent comprehensive review on this topic highlighted what the recent literature has provided in terms of the metabolite-based biomarkers which are useful for predicting and diagnosing DSM and for monitoring therapeutic interventions [
31]. One main limitation in understanding metabolic pathophysiology from the results of these studies is their limited sample sizes, which prevent the reaching of a definitive conclusion about the practicality of the identified DCM biomarkers for clinical purposes. Moreover, since these studies have mainly been conducted on biofluids such as serum, the alterations in plasma metabolites may represent the impact of the contribution of several organs. To overcome this last issue, a recent analysis by Flam et al. was conducted on myocardial biopsies from patients with end-stage HF; the analysis comprised metabolomics, genome-wide RNA sequencing, and global proteomic assessment [
32]. The findings confirmed the significant alterations in the metabolic process of the heart that had been seen previously in HF experiments with animal subjects, including a decline in the utilization of fatty acids and a heightened dependence on the utilization of ketones and carbohydrates. These results aligned with prior studies on human and animal models of HF. However, this particular study provides a novel insight. The previous research on animal models suggested that the decrease in fatty acid oxidation was due to a decline in mitochondrial oxidative function. However, the present study revealed a scarcity of fatty acids or acylcarnitines in the damaged heart, rather than an accumulation, indicating a shortage of fatty acid supply to the heart. As plasma fatty acid levels in HF patients remain unchanged, this scarcity in heart tissue could suggest a potential issue with fatty acid import. The specific cause of this is still unknown. This discovery is significant because it provides a new avenue for the targeting of the fatty acid metabolism in the treatment of HF. It is essential to determine whether similar changes occur during the development of HF and not just as a result of advanced disease. The study found that multiple classes of carbon substrates, including many amino acids, tricarboxylic acid cycle (TCA) metabolites, and glycolytic intermediates, were all reduced in the failing heart samples. This raises the possibility that these changes may reflect a state of malnutrition in end-stage disease and must be ruled out. Another difference between the human and animal studies is that HF patients often receive extensive treatment, which may impact the cardiac metabolism.
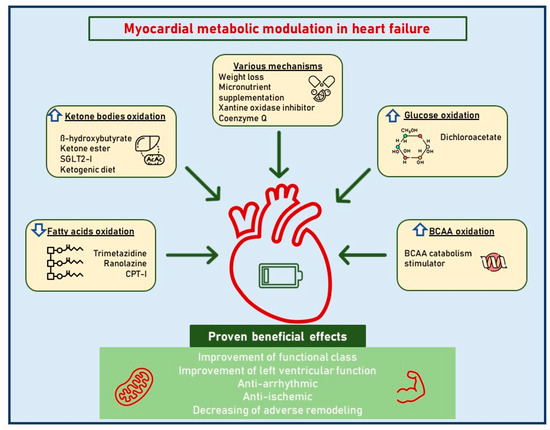
A better knowledge of cardiac metabolic adaptations in HF would certainly serve as a starting point from which to highlight new therapeutic targets, for old and new drugs, to exploit in different phenotypes and disease stages (Figure 1).
Figure 1. Myocardial metabolic modulation in dilated cardiomyopathy. This figure illustrates the different therapeutic metabolic targets in dilated cardiomyopathy. Abbreviations: SGLT2i, sodium-glucose cotransporter-2 inhibitors; BCAAs, branched-chain amino acids.
2. Metabolic Effects of Neurohormonal Hyperactivation Antagonists
2.1. Betablockers
Without affecting their detrimental chronotropic and inotropic effects, betablockers have the ability to directly alter myocardial energetics [
35]. By reducing peripheral lipolysis, this pharmacological class lowers the levels of FFA in the blood and allows a change in the heart’s energy metabolism that increases the use of carbohydrates [
36]. Through these metabolic processes, betablockers are responsible for this substrate competition’s decreased myocardial FFA uptake and increased glucose utilization [
37,
38]. A decrease in FFA delivery and an increase in the availability of arterial glutamate, which is highly advantageous for myocardial tissue as it can serve as both aerobic and anaerobic fuel, making it a particularly versatile substrate [
38], are both likely to be responsible for the increase in carbohydrate metabolism in the heart that was observed after a beta-blockade [
39,
40]. As a result, a beta-blockade may result in higher glucose consumption, which in turn may cause the heart to produce more energy without using up more oxygen. This suggests that in addition to their hemodynamic effects, betablockers may also directly affect the metabolic alterations observed in heart failure. When betablockers are used to treat systolic HF, greater energy efficiency and decreased oxygen consumption are seen. The changes in the way the heart produces energy may be the cause of these changes [
41]. In actuality, the phenomenon of heart-rate reduction in HF patients may only be a marker of a greater functional response to betablocker therapy [
42]. To lessen the failing heart’s reliance on fatty acids and to overcome the inhibition of myocardial glucose utilization brought on by fatty acids, the primary goal of therapy may be to reduce the plasma levels of FFAs and triacylglycerols. According to two studies, patients with New York Heart Association (NYHA) functional class III HF who took the betablocker carvedilol had lower FFA utilization and higher glucose utilization [
43,
44]. The reduction in oxygen consumption and the increase in energy effectiveness seen in HF patients after betablocker medication could possibly be explained by a change in the way the heart produces energy. In shifting the body’s energy substrate usage from lipid to glucose oxidation, non-selective betablockers appear to be more successful than selective ones [
44]. Non-selective betablockers, on the other hand, appear to worsen insulin resistance, which is already known to be linked to HF and CV disorders [
45,
46]. However, it does not seem that vasodilators and cardio-selective drugs promote insulin resistance. Carvedilol, in particular, may have beneficial metabolic effects on boosting insulin sensitivity in HF patients [
47]. Notably, the former’s greater metabolic effects may be one of the causes of the higher survival rates seen during their usage [
48].
2.2. RAAS Inhibitors
The hormone system known as RAAS controls blood pressure and fluid balance. Angiotensin I (AT I) conversion to angiotensin II (AT II) is blocked by RAAS inhibitors, and the AT II receptors at the end of the route are also blocked. Due to its direct ability to cause and sustain ventricular dysfunction through a variety of pathways, AT II is a key participant in the regulation of cardiac energy metabolism [
49]. It affects mitochondrial oxidative phosphorylation, particularly fatty acid oxidation [
50], and damages cardiomyocyte mitochondria by increasing the formation of reactive oxygen species [
51]. Additionally, there is proof that AT II reduces glucose oxidation [
52]. Overall, AT II can lower ATP levels by reducing oxidative metabolism [
53]. Its antagonism represents an appealing therapeutic strategy in this situation. Studies using the euglycemic insulin clamp technique demonstrated that the positive impact of AT II antagonistic action is exerted on insulin sensitivity. In fact, it has been demonstrated that ACE inhibitors [
54] and angiotensin receptor antagonists [
55] improve glucose homeostasis and left ventricular performance. The potential routes of action include elevated skeletal muscle blood flow, bradykinin build up, or more effective insulin release. Finally, RAAS inhibitors are able to reduce the atrial wall stress and fibrosis, consequently promoting a progressive reverse remodeling of the enlarged left atrium in dilated cardiomyopathy, with positive outcome effects [
56].
2.3. Angiotensin Receptor Neprilysin Inhibitors (ARNI)
After a median follow-up of 27 months, the PARADIGM-HF study demonstrated that sacubitril/valsartan, the first member of a new class of medications known as ARNI, reduced the morbidity and mortality of patients with HF and reduced EF compared with the ACE inhibitor enalapril [
57]. Sacubitril/valsartan is thought to provide an extra benefit over the renin–angiotensin blockade alone because it inhibits neprilysin, an endopeptidase that breaks down endogenous vasoactive peptides such natriuretic peptides [
58]. Although there were very few patients who developed new-onset diabetes during the course of the PARADIGM-HF trial, sacubitril/valsartan did not lower the pre-specified exploratory outcome of new-onset diabetes, in contrast to enalapril. Despite this, new research suggests that sacubitril/valsartan, regardless of diabetes, may enhance lipid metabolism, insulin sensitivity, and glucose metabolism in individuals with HF [
59,
60].
2.4. Mineralocorticoid Receptor Antagonists (MRAs)
Aldosterone has negative effects that are mediated via the mineralocorticoid receptor; these effects are blocked by MRAs such as spironolactone and eplerenone. MRAs are therefore effective in treating hypertension, especially resistant hypertension, and in lowering the risk of morbidity and death in HF patients through this pharmacological activity. The “off-target effects” of spironolactone have also been shown to have negative effects on lipid and glucose homeostasis [
61]. The blockage of glucocorticoid receptors by spironolactone is thought to be the mechanism by which cortisol blood concentrations are raised. By accelerating lipolysis and gluconeogenesis, the glucocorticoid cortisol raises blood glucose levels. On the other hand, the selective MRA eplerenone has relatively little effect on other steroid receptors. Because of this, it does not impact glucose metabolism, and it lowers serum cortisol levels [
61]. It has been confirmed that spironolactone may cause changes in blood glucose levels, whereas eplerenone has no effect on glucose homeostasis, according to a recent systematic review of randomized controlled trials, prospectives, and observational studies evaluating the influence of the various MRAs on the biomarkers of glucose homeostasis in a variety of populations [
62,
63]. The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF) study of participants with chronic HF actually had no effect on new-onset diabetes [
64].
2.5. Loop Diuretics
Loop diuretics, such as furosemide, primarily affect the kidneys to increase urine production. However, they may also have an indirect effect on the heart’s metabolism. Hypomagnesemia and hypokalemia can be brought on by loop diuretics. Arrhythmias are more likely to occur in people with these two conditions, which can impair cardiac function by interfering with normal electrical signaling. Additionally, hypocalcemia, which may affect cardiac contractility, can be brought on by these medications. Last but not least, loop diuretics can contribute to all of the metabolic changes associated with RAAS activation (as previously described) by stimulating the RAAS through volume depletion and decreased blood pressure [
65].
3. Direct Cardiac Metabolism Modulators
Although the ideal metabolic environment for a failing heart remains highly debated and poorly understood, it may be a good target for future treatments. With the use of medications such as trimetazidine, ranolazine, perhexiline, and etomoxir, the previous research has concentrated on boosting glucose oxidation, which, in comparison to FFA oxidation, has a higher P/O ratio. These medications have been shown to enhance myocardial energetics and LV function in patients with chronic ischemic and non-ischemic HF and low EF. For unknown reasons, these medications have not, however, ever undergone extensive clinical studies [
66].
The failing heart in severe non-ischemic cardiomyopathy, on the other hand, may be metabolically adaptive and not oxygen-restricted, according to some preliminary investigations. This challenges the conventional wisdom that the optimal therapeutic goal is to increase glucose oxidation; instead, it supports the alternative position that increasing fatty acid oxidation is a wise course of action [
66].
4. The Ketone Bodies Hypothesis
As outlined above, targeting cardiac metabolism by decreasing fatty acid oxidation and promoting glucose oxidation appears to be an interesting approach to the treatment of chronic HF. Several drugs have been investigated in small-scale studies, but large clinical trials are needed to confirm the efficacy of these agents as a part of chronic HF treatment.
More recently, a new interest in ketone body metabolism has arisen as their modulation may be of potential benefit to HF patients.
Under normal conditions, ketones represent a minimal part of all substrates utilized by the myocardium for energy production. These compounds, however, become critical during periods of stress and fasting since their utilization allows the preservation of glycogen stores. The myocardium is the highest ketone body consumer per unit mass. Ketone body oxidation is also more efficient than fatty acid oxidation in terms of ATP synthesis per molecule of oxygen used [
111,
112]. In addition, ketone body metabolism exerts anti-oxidant effects since it oxidizes mitochondrial co-enzyme Q and reduces cytosolic [NADP+]/[NADPH+], thereby decreasing free radical production [
112]. The resourcefulness of the heart in using ketone bodies in order to satisfy its ATP requirements serves as a tool to spare glucose. However, it remains rather unclear whether their employment is compensative to balance out the negative effects of the failing heart adaptive/maladaptive substrate utilization.
In hypertrophied and early-stage failing rat hearts, a reduced capacity to oxidize fatty acids and a shift to ketone oxidation as an alternative metabolic fuel have been observed [
113]. Similar data have been found in failing human hearts: patients with reduced LV EF nearly tripled their consumptions of ketones as metabolic substrates compared to patients with preserved EF [
27].
A case control study involving patients with chronic dilated non-ischemic cardiomyopathy showed increased amounts of beta-hydroxybutyryl CoA and decreased amounts of myocardial beta-hydroxybutyrate in myocardial tissue, suggesting an increased ketone body metabolism in this setting [
114].
Additional studies have shown that circulating ketone bodies in subjects with chronic HF increase proportionally to the intensity of their symptoms, the level of congestion in the venous circulatory system, and the magnitude of neurohormonal and cytokine involvement, as well as the increasing deterioration of left ventricular function [
115,
116]. In this context, more ketones are produced through hepatic ketogenesis and become a fundamental substrate for energy production in cardiomyocytes [
117].
These results clearly indicate that chronic HF determines a ketosis-prone state [
115]. Indeed, exhaled acetone levels have been shown to be able to identify HF patients with a predictive value which is somewhat similar to that of brain natriuretic peptide (BNP); moreover, this predictive value is proportional to the NYHA class [
116].
It is also known that exhaled breath acetone is increased in HF patients with reduced EF and is associated with higher mortality or heart transplantation [
118].
Interestingly, higher serum levels of beta-hydroxybutyrate seem to relate to disease progression and adverse prognosis in arrhythmogenic cardiomyopathy patients, supporting the hypothesis that an enhanced ketone body metabolism may be a standard myocardium response to injuries [
119].
According to another study, the cardiomyocytes’ specific loss of succinyl-CoA:3-oxoacid CoA transferase, which is involved in ketone body oxidation, is associated with significantly increased left ventricular volume and a decreased ejection fraction as a response to pressure overload [
120]. Overall, these studies confirm the fundamental role of this metabolic pathway, showing that impaired ketone body oxidation may be associated with worsened heart remodeling following pressure overload.
In this context, the concept of the therapeutic modulation of ketone metabolism as a potential new target in HF treatment is emerging [
120,
121,
122,
123,
124].
5. Sodium-Glucose Cotransporter-2 Inhibitors (SGLT2i)
There is a bidirectional link between diabetes mellitus (DM) and HF. Longstanding diabetes causes changes in myocardial metabolism, abnormal calcium signaling, and inflammatory pathways, resulting in structural and functional changes in the myocardium and leading to the development of diabetic cardiomyopathy and the progression of HF [
125,
126]. Conversely, HF patients without DM are at an increased risk of developing glycemic abnormalities [
125]. The shared underlying risk factors and the overlap of the pathophysiological mechanisms play a critical role in the frequent coexistence of DM and HF. As with HF, there is also a strong link between diabetes, coronary artery disease, hypertension, and renal disease.
During the last decade, cardiovascular outcome trials have investigated several classes of new glucose-lowering agents, including SGLT2i, which, apart from showing evidence of cardiovascular safety, have also been shown to exert beneficial effects on the cardiovascular outcome [
127,
128]. Most studies have shown the independence of cardiovascular outcome from glycemic control, indicating mechanisms of action other than those usually postulated to explain the cardiovascular benefits of glucose-lowering therapies [
129,
130,
131,
132,
133,
134]. In fact, the significant beneficial clinical effects observed with SGLT2i use cannot be explained by one single mechanism.
This entry is adapted from the peer-reviewed paper 10.3390/jcdd10070287