Tissue plasminogen activator (tPA) is a serine protease regulating the homeostasis of blood coagulation, fibrinolysis, and matrix degradation, and has been shown to act as a cytokine to trigger various receptor-mediated intracellular signal pathways, modulating macrophage function in response to kidney injury.
- tissue plasminogen activator (tPA)
- macrophage function
- signal transduction
- inflammation
- nuclear factor- κB (NF-κB)
- low-density lipoprotein receptor-related protein (LRP-1)
- annexin A2
- macrophage polarization
1. Introduction
2. tPA Structure and Dual Functions of Protease and Cytokine
3. Renal Origin and Distribution of tPA after Kidney Injury
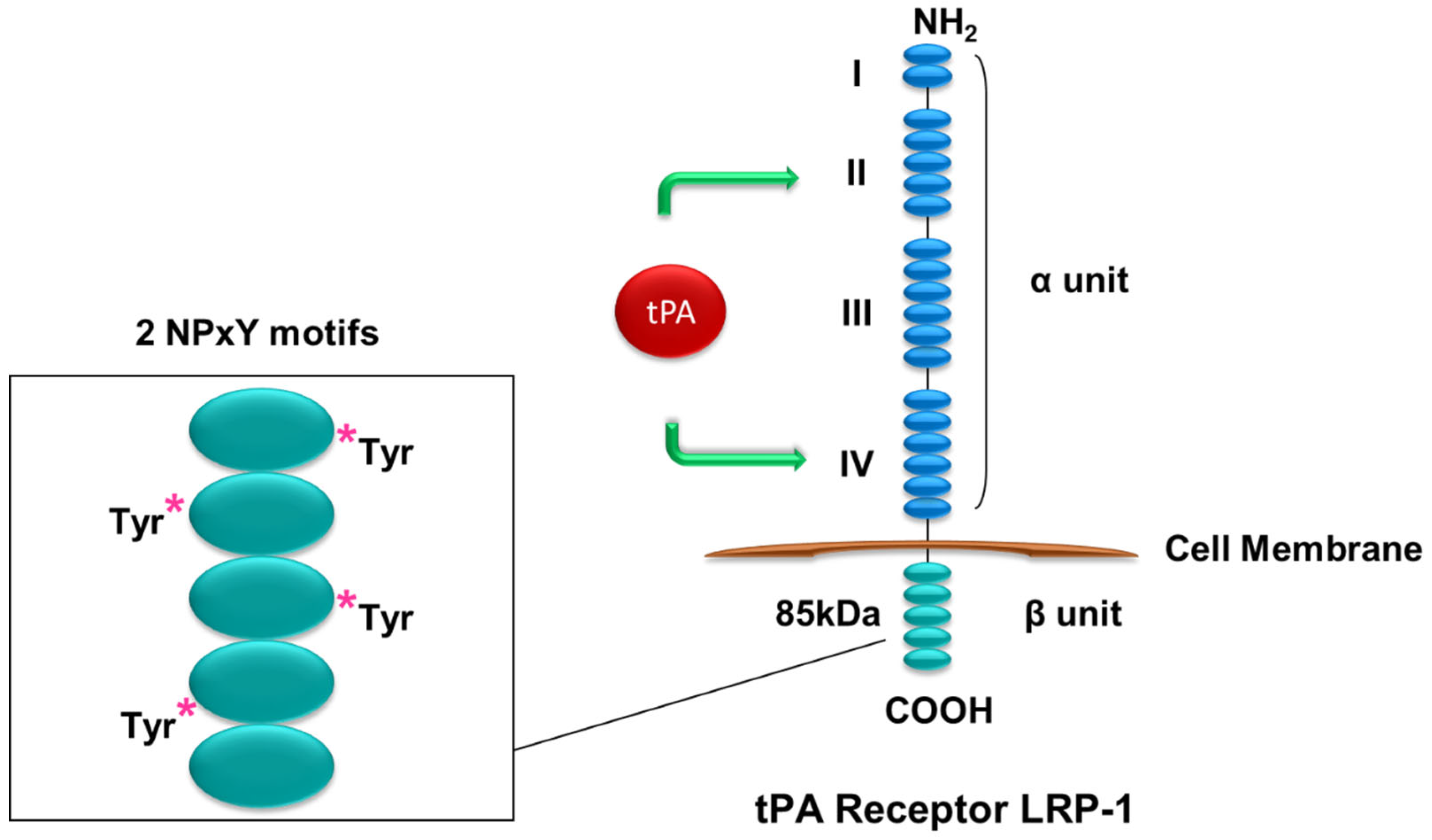
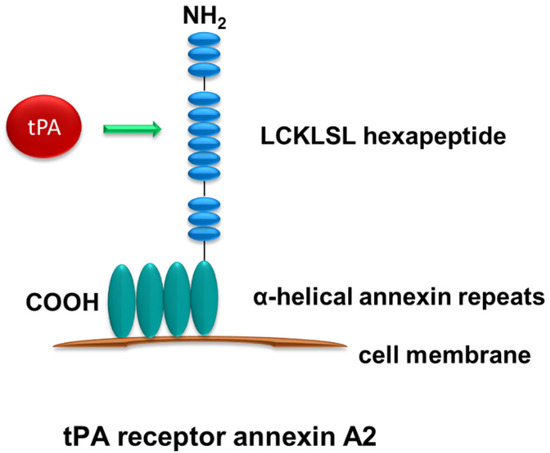
4. Renal Receptors of tPA Signaling
5. tPA and NF-kB Signaling in Renal Inflammation
The researchers have hypothesized that tPA promotes renal inflammation through modulating NF-κB pathway based on the previous findings of the concurrent induction of tPA and activation of NF-κB during the progression of CKD and the competency of tPA in regulating renal inflammatory responses [29]. The researchers have investigated the role of tPA and NF-κB in a mouse UUO model of CKD and have found that tPA promotes kidney fibrosis and inflammation. After obstructive injury, tPA knockout mice have displayed less collagen deposition, fewer CD11b-positive macrophage infiltration, and reduced activation of NF-kB in the diseased kidneys, as indicated by lower renal p65 phosphorylation and less NF-kB-dependent chemokines. The researchers have further demonstrated that tPA activates NF-κB signaling independent of its protease activity via the canonical pathway in macrophages by inducing IκB phosphorylation and triggering the nuclear translocation of p65/RelA, and subsequently transcription of NF-kB-dependent chemokines such as interferon-γ-inducible protein (IP)-10 and macrophage inflammatory protein (MIP)-1 α [16]. Intriguingly, LRP-1 as the most-studied receptor of tPA does not mediate tPA-activated NF-κB signaling, because siRNA knockdown of LRP-1 has had little effects. Instead, annexin A2 has been shown to mediate tPA’s effects. Because cell surface annexin A2 lacks the transmembrane domain and can only dock onto cell membrane in a peripheral manner [37], the researchers have proposed that annexin A2 may function as a coreceptor of a known outside-in signal transduction pathway, such as the integrin signaling. It’s known that tPA binds to integrin Mac-1 (CD11b/CD18) in macrophages [23], the researchers have investigated the possible interaction between annexin A2 and CD11b. It has been discovered that tPA promotes the aggregation of annexin A2 and CD11b in macrophages, and such interaction is also induced in the obstruction-injured kidney. Further in vitro studies have defined that tPA-induced aggregation of annexin A2 and CD11b activates its immediate downstream effector integrin-linked kinase (ILK), which, in turn, leads to phosphorylation and degradation of IκB, release and nuclear translocation of NF-κB dimers, and subsequently DNA binding and transcription of target proinflammatory genes [16]. These findings have defined a novel signaling mechanism of tPA-mediated NF-κB activation in promoting macrophage infiltration and renal inflammation. It’s worth mentioning that macrophages also produce tPA which may activate NF-κB signaling in macrophages in an autocrine manner initiating a vicious cycle of amplification.
Of note, it should be pointed out that the effect of tPA on NF-κB signaling is context dependent and affected by many factors, such as cell types, cellular states, and organ specificities. One of the notable examples is that tPA has been shown to suppress NF-κB activation in LPS-stimulated macrophages through LRP-1-mediated miR-155 [56]. The anti-inflammatory activity of tPA is mediated through the interaction of LRP-1 and its coreceptor N-methyl-D-aspartate Receptor (NMDA-R), which is characterized as a neuron ionotropic glutamate receptor in neurons, macrophages and Schwann cells [56][57]. LRP-1/NMDA-R-mediated effect is cellular state-dependent [58]. Non-stimulated mouse peritoneal macrophages express very low levels of the NMDA-R and are not responsive to tPA. However, after treatment with colony-stimulating factor-1 (CSF-1), activated macrophages display increased cell-surface NMDA-R and responsiveness to tPA [59]. Moreover, Zhang et al. has demonstrated that tPA and LRP-1 mediates cerebral ischemia-induced NF-κB activation [18]. They have found that ischemic insult-induced NF-κB activation is attenuated after LRP inhibition or tPA knockout [18]. Therefore, tPA may execute cell type-specific biological functions by binding to different membrane receptors (LRP-1 or annexin A2) or their coreceptor, such as NMDA-R and CD11b, and initiating different intracellular signaling events to modulate NF-κB activation [16][18][56].
This entry is adapted from the peer-reviewed paper 10.3390/ijms241311067
References
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661.
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530.
- Lin, L.; Hu, K. Tissue-type plasminogen activator modulates macrophage M2 to M1 phenotypic change through annexin A2-mediated NF-kappaB pathway. Oncotarget 2017, 8, 88094–88103.
- Ferrario, F.; Castiglione, A.; Colasanti, G.; Barbiano di Belgioioso, G.; Bertoli, S.; D’Amico, G. The detection of monocytes in human glomerulonephritis. Kidney Int. 1985, 28, 513–519.
- Eardley, K.S.; Kubal, C.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Savage, C.O.; Howie, A.J.; Kaur, K.; Cooper, M.S.; Adu, D.; et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008, 74, 495–504.
- Hu, K.; Mars, W.M.; Liu, Y. Novel actions of tissue-type plasminogen activator in chronic kidney disease. Front. Biosci. 2008, 13, 5174–5186.
- Vivien, D.; Gauberti, M.; Montagne, A.; Defer, G.; Touze, E. Impact of tissue plasminogen activator on the neurovascular unit: From clinical data to experimental evidence. J. Cereb. Blood Flow. Metab. 2011, 31, 2119–2134.
- Olson, S.T.; Swanson, R.; Day, D.; Verhamme, I.; Kvassman, J.; Shore, J.D. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry 2001, 40, 11742–11756.
- White, S.; Lin, L.; Hu, K. NF-kappaB and tPA Signaling in Kidney and Other Diseases. Cells 2020, 9, 1348.
- Hu, K.; Lin, L.; Tan, X.; Yang, J.; Bu, G.; Mars, W.M.; Liu, Y. tPA protects renal interstitial fibroblasts and myofibroblasts from apoptosis. J. Am. Soc. Nephrol. 2008, 19, 503–514.
- Hu, K.; Wu, C.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J. Clin. Investig. 2007, 117, 3821–3832.
- Hu, K.; Yang, J.; Tanaka, S.; Gonias, S.L.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J. Biol. Chem. 2006, 281, 2120–2127.
- Lin, L.; Bu, G.; Mars, W.M.; Reeves, W.B.; Tanaka, S.; Hu, K. tPA activates LDL receptor-related protein 1-mediated mitogenic signaling involving the p90RSK and GSK3beta pathway. Am. J. Pathol. 2010, 177, 1687–1696.
- Lin, L.; Wu, C.; Hu, K. Tissue Plasminogen Activator Activates NF-kappaB through a Pathway Involving Annexin A2/CD11b and Integrin-Linked Kinase. J. Am. Soc. Nephrol. 2012, 23, 1329–1338.
- Higazi, A.A.; El-Haj, M.; Melhem, A.; Horani, A.; Pappo, O.; Alvarez, C.E.; Muhanna, N.; Friedman, S.L.; Safadi, R. Immunomodulatory effects of plasminogen activators on hepatic fibrogenesis. Clin. Exp. Immunol. 2008, 152, 163–173.
- Zhang, X.; Polavarapu, R.; She, H.; Mao, Z.; Yepes, M. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am. J. Pathol. 2007, 171, 1281–1290.
- Huber, D.; Cramer, E.M.; Kaufmann, J.E.; Meda, P.; Masse, J.M.; Kruithof, E.K.; Vischer, U.M. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood 2002, 99, 3637–3645.
- Yepes, M.; Woo, Y.; Martin-Jimenez, C. Plasminogen Activators in Neurovascular and Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4380.
- Roelofs, J.J.; Rouschop, K.M.; Leemans, J.C.; Claessen, N.; de Boer, A.M.; Frederiks, W.M.; Lijnen, H.R.; Weening, J.J.; Florquin, S. Tissue-type plasminogen activator modulates inflammatory responses and renal function in ischemia reperfusion injury. J. Am. Soc. Nephrol. 2006, 17, 131–140.
- Yang, J.; Shultz, R.W.; Mars, W.M.; Wegner, R.E.; Li, Y.; Dai, C.; Nejak, K.; Liu, Y. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J. Clin. Investig. 2002, 110, 1525–1538.
- Cao, C.; Lawrence, D.A.; Li, Y.; Von Arnim, C.A.; Herz, J.; Su, E.J.; Makarova, A.; Hyman, B.T.; Strickland, D.K.; Zhang, L. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006, 25, 1860–1870.
- Lin, L.; Jin, Y.; Mars, W.M.; Reeves, W.B.; Hu, K. Myeloid-derived tissue-type plasminogen activator promotes macrophage motility through FAK, Rac1, and NF-kappaB pathways. Am. J. Pathol. 2014, 184, 2757–2767.
- Bu, G.; Williams, S.; Strickland, D.K.; Schwartz, A.L. Low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor is an hepatic receptor for tissue-type plasminogen activator. Proc. Natl. Acad. Sci. USA 1992, 89, 7427–7431.
- Herz, J.; Strickland, D.K. LRP: A multifunctional scavenger and signaling receptor. J. Clin. Investig. 2001, 108, 779–784.
- Hussain, M.M. Structural, biochemical and signaling properties of the low-density lipoprotein receptor gene family. Front. Biosci. 2001, 6, D417–D428.
- Strickland, D.K.; Ranganathan, S. Diverse role of LDL receptor-related protein in the clearance of proteases and in signaling. J. Thromb. Haemost. 2003, 1, 1663–1670.
- Lin, L.; Hu, K. Tissue plasminogen activator and inflammation: From phenotype to signaling mechanisms. Am. J. Clin. Exp. Immunol. 2014, 3, 30–36.
- Obermoeller-McCormick, L.M.; Li, Y.; Osaka, H.; FitzGerald, D.J.; Schwartz, A.L.; Bu, G. Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J. Cell. Sci. 2001, 114, 899–908.
- Barnes, H.; Ackermann, E.J.; van der Geer, P. v-Src induces Shc binding to tyrosine 63 in the cytoplasmic domain of the LDL receptor-related protein 1. Oncogene 2003, 22, 3589–3597.
- Barnes, H.; Larsen, B.; Tyers, M.; van Der Geer, P. Tyrosine-phosphorylated low density lipoprotein receptor-related protein 1 (Lrp1) associates with the adaptor protein SHC in SRC-transformed cells. J. Biol. Chem. 2001, 276, 19119–19125.
- Strickland, D.K.; Gonias, S.L.; Argraves, W.S. Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 2002, 13, 66–74.
- Loukinova, E.; Ranganathan, S.; Kuznetsov, S.; Gorlatova, N.; Migliorini, M.M.; Loukinov, D.; Ulery, P.G.; Mikhailenko, I.; Lawrence, D.A.; Strickland, D.K. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function betwenn LRP and the PDGF. J. Biol. Chem. 2002, 277, 15499–15506.
- Lin, L.; Hu, K. Annexin A2 and Kidney Diseases. Front. Cell. Dev. Biol. 2022, 10, 974381.
- Siao, C.J.; Tsirka, S.E. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J. Neurosci. 2002, 22, 3352–3358.
- Rescher, U.; Gerke, V. Annexins--unique membrane binding proteins with diverse functions. J. Cell. Sci. 2004, 117, 2631–2639.
- Hajjar, K.A.; Mauri, L.; Jacovina, A.T.; Zhong, F.; Mirza, U.A.; Padovan, J.C.; Chait, B.T. Tissue plasminogen activator binding to the annexin II tail domain. Direct modulation by homocysteine. J. Biol. Chem. 1998, 273, 9987–9993.
- Kim, J.; Hajjar, K.A. Annexin II: A plasminogen-plasminogen activator co-receptor. Front. Biosci. 2002, 7, d341–d348.
- Cesarman, G.M.; Guevara, C.A.; Hajjar, K.A. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J. Biol. Chem. 1994, 269, 21198–21203.
- Ortiz-Zapater, E.; Peiro, S.; Roda, O.; Corominas, J.M.; Aguilar, S.; Ampurdanes, C.; Real, F.X.; Navarro, P. Tissue plasminogen activator induces pancreatic cancer cell proliferation by a non-catalytic mechanism that requires extracellular signal-regulated kinase 1/2 activation through epidermal growth factor receptor and annexin A2. Am. J. Pathol. 2007, 170, 1573–1584.
- Sharma, M.; Ownbey, R.T.; Sharma, M.C. Breast cancer cell surface annexin II induces cell migration and neoangiogenesis via tPA dependent plasmin generation. Exp. Mol. Pathol. 2010, 88, 278–286.
- Paciucci, R.; Tora, M.; Diaz, V.M.; Real, F.X. The plasminogen activator system in pancreas cancer: Role of t-PA in the invasive potential in vitro. Oncogene 1998, 16, 625–633.
- Bonizzi, G.; Karin, M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288.
- Sanz, A.B.; Sanchez-Nino, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262.
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034.
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260.
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558.
- Sterner, R.M.; Hartono, S.P.; Grande, J.P. The Pathogenesis of Lupus Nephritis. J. Clin. Cell. Immunol. 2014, 5, 1357–1366.
- Kiryluk, K.; Novak, J. The genetics and immunobiology of IgA nephropathy. J. Clin. Investig. 2014, 124, 2325–2332.
- Guijarro, C.; Egido, J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 2001, 59, 415–424.
- Zhang, H.; Sun, S.C. NF-kappaB in inflammation and renal diseases. Cell. Biosci. 2015, 5, 63.
- Morrissey, J.; Klahr, S. Transcription factor NF-kappaB regulation of renal fibrosis during ureteral obstruction. Semin. Nephrol. 1998, 18, 603–611.
- Volpini, R.A.; Costa, R.S.; da Silva, C.G.; Coimbra, T.M. Inhibition of nuclear factor-kappaB activation attenuates tubulointerstitial nephritis induced by gentamicin. Nephron Physiol. 2004, 98, p97–p106.
- Fujihara, C.K.; Antunes, G.R.; Mattar, A.L.; Malheiros, D.M.; Vieira, J.M., Jr.; Zatz, R. Chronic inhibition of nuclear factor-kappaB attenuates renal injury in the 5/6 renal ablation model. Am. J. Physiol. Ren. Physiol. 2007, 292, F92–F99.
- Gonias, S.L. Plasminogen activator receptor assemblies in cell signaling, innate immunity, and inflammation. Am. J. Physiol. Cell. Physiol. 2021, 321, C721–C734.
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39.
- Mantuano, E.; Azmoon, P.; Brifault, C.; Banki, M.A.; Gilder, A.S.; Campana, W.M.; Gonias, S.L. Tissue-type plasminogen activator regulates macrophage activation and innate immunity. Blood 2017, 130, 1364–1374.
- Das, L.; Azmoon, P.; Banki, M.A.; Mantuano, E.; Gonias, S.L. Tissue-type plasminogen activator selectively inhibits multiple toll-like receptors in CSF-1-differentiated macrophages. PLoS ONE 2019, 14, e0224738.