Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Polymer Science
Organocatalysis, the use of chiral organic molecules as catalysts, has emerged as a highly efficient alternative to traditional asymmetric catalysis methods. Chiral porous organic frameworks have emerged as candidates for heterogeneous asymmetric organocatalysis.
- covalent organic frameworks
- covalent triazine frameworks
- conjugated microporous polymers
- asymmetric organocatalysis
- chirality
1. Introduction
Chirality has important implications for many biological processes and plays a crucial role in the development of new drugs and materials [1,2]. For instance, more than half of the commercial pharmaceuticals are chiral. Thus, in the recent years, a growing interest in enantiomerically pure compounds has emerged in medicinal chemistry because of the possible toxicity of inactive enantiomers [3,4]. In addition, chirality is relevant for a wide variety of other fields, such as crop protection, flavors, fragrancies, and synthetic chemistry [5,6,7,8]. Therefore, asymmetric catalysis, the use of chiral catalysts to selectively produce a desired chiral product, is an important research field that includes several strategies, such as the use of metal complexes or enzymes.
Organocatalysis, the use of chiral organic molecules as catalysts, has emerged as a highly efficient alternative to traditional asymmetric catalysis methods. Chiral organocatalysts correspond to different kinds of molecules such as amines or amino acids, Brønsted acids, phosphoric acids, or imidazolidinones [9]. Asymmetric organocatalysis offers several advantages over traditional strategies. For instance, chiral organic molecules are generally less toxic and more environmentally friendly than metal catalysts [10]. Furthermore, chiral organic molecules can be readily synthesized and are often less expensive. Asymmetric organocatalysis also offers high functional group compatibility, as the reactions can be carried out under mild conditions, reducing the risk of unwanted side reactions. However, organocatalysis suffers from some general drawbacks, such as the need for high catalyst loading, low catalyst stability, and the difficulty in catalyst recovery [11].
Owing to the inherent advantages of asymmetric organocatalysis, the research on systems that allow to overcome their common drawbacks, described above, is a major goal in modern chemistry. To this end, the incorporation of organocatalytic fragments into porous materials is a strategy that has recently started to blossom [12]. A particularly appealing family of porous frameworks with a great potential are those constructed by the covalent assembly of exclusively organic building blocks. Such materials, covalent organic frameworks and their amorphous analogs, offer several advantages over other types of porous materials [13,14]. First, porous organic materials can present high stability under a wide range of conditions, including high temperatures and acidic or basic environments. Moreover, they possess an extraordinarily tailorable structures and, therefore, their properties can be precisely tuned by modifying their molecular precursors, which can include chiral moieties of different nature [15]. Thus, in the literature a growing number of examples of covalent organic frameworks containing chiral fragments have started to appear, as well as their non-crystalline counterparts.
2. Critical Considerations on the Classification of Porous Organic Frameworks
Porous organic frameworks are materials designed according to the reticular strategy by connecting predetermined building blocks to generate predictable structures and topologies. The term reticular refers to the structure of the materials obtained, which consists of a network of nodes and linkers that form a three-dimensional framework, and can arise from the expansion of a 3D geometry or from the ordered staking of layered structures [16]. Strictly, the definition reticular chemistry implies the isolation of crystalline materials, which is not always the case for the extended structures included herein. In fact, researchers have included catalytic applications of crystalline and amorphous structures.
The goal of reticular design is to create materials with tailored properties, such as specific pore sizes, shapes, and pore surface functionalities, which can be used for a variety of applications, including catalysis. As a result of their potential advanced catalytic applications, these materials could address some of the most urgent global social challenges, such as energy and environmental sustainability, and are, therefore, the subject of intense research worldwide [17].
A particular research playground is the synthetic design and application of porous organic frameworks. One of the most attractive features of these materials is the fact that they are constructed from interconnected networks of organic molecules and, as a consequence, possess a high degree of tunability. Therefore, it is possible to design and synthesize extended organic materials with a wide range of different properties, making them suitable for use in a variety of different contexts.
The assembly of organic materials following reticular design has resulted in a plethora of materials that correspond to different denominations. The first and most popular family of reticular organic materials are the Covalent Organic Frameworks (COFs), which are defined as follows: “Class of materials that form two- or three-dimensional structures through reactions between organic precursors resulting in strong, covalent bonds to afford porous, stable, and crystalline materials” [18]. Depending on the geometry of the selected building blocks, the extended structures can adopt several laminar or 3D topologies. This structural design gives rise to engineered pores of predictable sizes and shapes, as shown in Figure 1. The most common approaches for synthesizing COFs include solvothermal, solid-state, microwave-assisted methods, and condensation reactions performed at room temperature [19]. In all cases, the reactivity between the functional groups on the organic building blocks leads to the formation of strong covalent bonds as links of the extended organic framework. The types of linkages that have been used for the preparation of COFs include, among others, boronic esters, boroxine, imine, hydrazone, phenazine, azine, imide, and triazine moieties (Scheme 1) [20,21].

Figure 1. Representative topologies commonly obtained in the assembly COFs.
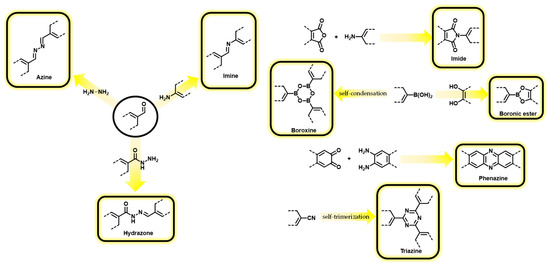
Scheme 1. Reactions commonly used in the assembly of COFs.
A key feature that is inherent to the COF definition is crystallinity, allowing to assign these materials the tag of “reticular chemistry”. This feature is highly relevant for certain applications, such as gas storage and separation, because it is associated with a highly ordered structure with well-defined pores. However, achievement of this property is restricted to condensation reactions with a high degree of reversibility (Scheme 1) that can result, under specific reaction conditions, in the self-healing of structural defects and eventually to preserved long-range structural order.
Amorphous reticular organic designs have been classified in categories different from COFs. One family of amorphous porous organic frameworks are the so-called Conjugated Microporous Polymers (CMPs), which are defined as a class of organic materials that combine extended π-conjugation with a permanently microporous layered structure. The high degree of electron mobility in CMPs makes them very attractive for optoelectronic and photocatalytic applications. The porous structure and electronic properties of CMPs can be tailored by varying the monomer composition, degree of polymerization, and the reaction conditions. CMPs can be synthesized using various polymerization techniques, such as oxidative, Sonogashira, and palladium-catalyzed coupling reactions (Scheme 2) [22].
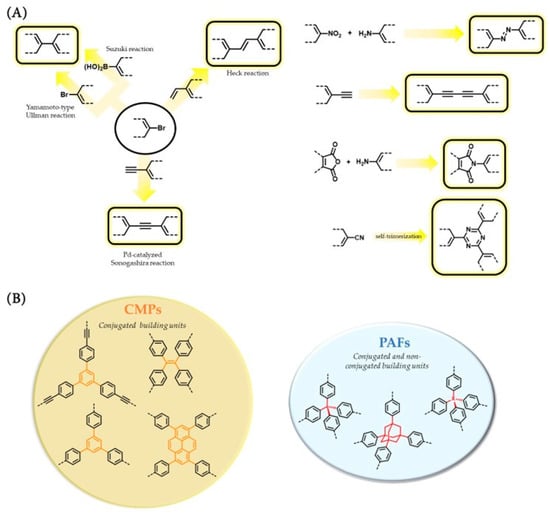
Scheme 2. (A) Reactions commonly used in the assembly of CMPs and PAFs. (B) Representative basic structural motifs in CMPs and PAFs.
A specific design of conjugated microporous materials is the one that corresponds to Covalent Triazine Frameworks (CTFs), which can be defined as a class of extended porous organic materials that are composed of triazine-based moieties linked by covalent bonds [23]. Triazine fragments in CTFs provide a flat (layered) structure with high stability. The aromatic carbon–nitrogen bond is very stable and irreversible under standard conditions, and thus, CTFs are generally highly chemically and thermally stable and amorphous in nature. The general synthetic methods to obtain CTFs consist of the cyclotrimerization reaction of the nitrile functional group using Brønsted acids, or Suzuki cross-coupling, Friedel–Crafts reaction, or amidine-mediated procedures [23]. Thus, although amorphous CTFs can be considered a subclass of CMPs because they are microporous and have extended π-conjugation, they are usually denominated according to their specific nomenclature because they were developed separately, and the chemistry of their formation was initially quite different. However, in some limited cases, CTFs synthesized through ionothermal or microwave-assisted methods present partially crystalline structures, which opens the door of the COF realm to CTFs [24,25].
A further twist in the field of reticular designs for organic materials was the appearance of materials known as Porous Aromatic Frameworks (PAFs), which are commonly defined as porous organic polymer formed exclusively by aromatic rigid linkers assembled through strong covalent bonds [26]. Interestingly, first PAF, PAF-1, resulted from the assembly of the tetrakis(4-bromophenyl)methane building block by means of Yamamoto-type Ullman cross-coupling reaction (Scheme 2) [27]. The resulting material was a 3D network containing biphenyl fragments held together by sp3-carbon atoms. Thereafter, many 3D PAFs were developed based on building blocks containing sp3-C atoms. Therefore, PAFs that do not possess extended π-conjugation (which is broken by tetrahedral sp3-C) should be considered as a class of porous organic frameworks different from that of CMPs. However, the definition of PAFs does not exclude laminar designs, which could result in an overlap with the CMP denomination.
Analysis of the available literature reveals a striking ambiguity on which structures could be considered CMPs or a laminar PAFs. For instance, the exactly same chemical structure can be found in literature as CTF-3 or PAF-56 [28,29]. Although the synthetic routes are significantly different, assigning different classifications to identical structures may lead to confusion. Another example of very similar structures consists of porphyrin units assembled through biphenyl (PAF-40) or terphenyl (FeP-CMP) fragments [30,31]. Despite their similarity, the structures are assigned to PAF and CMP families, respectively. Many other examples of laminar designs denominated as PAFs but undistinguishable from CMPs can be found in the literature. For instance, assembly via Sonogashira–Hagihara coupling reaction of 1,3,6,8-tetrabromopyrene with 1,4-diethynylbenzene or 1,3,5-triethynylbenzene resulted in conjugated structures with permanent porosities—therefore qualifying them as CMPs—albeit being denominated as PAF-19 and PAF-20, respectively [32].
Overall, considering the lack of clarity in the classification of amorphous COF counterparts (Figure 2), the researchers in the field need to establish unified criteria. Otherwise, further confusion will be added to the literature, increasing the difficulty in the search for published information. As a suitable guideline, researchers propose to make a clear difference between CMP and PAF materials (Figure 2). In this respect, considering the seminal works that reported such structures, it seems reasonable to restrict the PAF definition to non-conjugated structures, such as the 3D architectures that were initially reported under the PAF denomination. Otherwise, it appears logical to propose that conjugated materials should be exclusively denominated as CMPs. In a more general view, a common name should be found for all COFs and their amorphous analogs that are also designed using a reticular approach. Using terminology found in the literature, this comprehensive group of materials could be known as porous organic frameworks (POFs).

Figure 2. Current classification of extended organic materials. The representative examples of amorphous materials are shown.
3. Introduction of Chirality into Porous Organic Frameworks
There are four main strategies to incorporate chirality into porous organic frameworks (Figure 3):
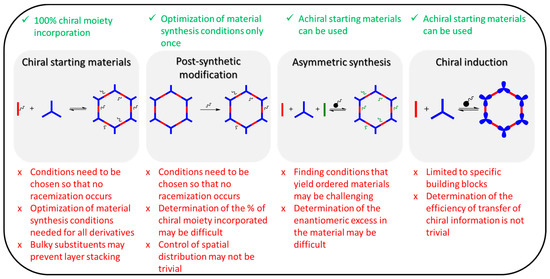
Figure 3. The reported approaches to incorporate chirality into reticular organic materials and related advantages and disadvantages (ticks denote advantages and X denotes disadvantages).
- (i)
-
Synthesis using chiral building blocks;
- (ii)
-
Post-synthetic modification;
- (iii)
-
Asymmetric synthesis;
- (iv)
-
External chiral induction.
The most straightforward approach is (i) using chiral building blocks as the starting materials, as this approach ensures the full incorporation of the chiral moieties into the material [33,34,35,36,37,38]. Typically, only one of the building blocks contains the chiral moiety. However, care needs to be taken that racemization of the building block does not occur under the chosen synthesis conditions. In addition, the reaction conditions need to be optimized separately for each building block. Furthermore, the use of bulky building blocks may prevent stacking interactions, which could hinder the formation of COF structures. This could potentially be mitigated using a mixed-linker strategy, where the building block bearing the chiral information is mixed with a less bulky non-chiral compound, although in such a case determining the number of chiral groups incorporated into the material may be challenging.
In (ii) post-synthetic modification (PSM), chiral moieties are incorporated to an already synthesized material [39,40,41,42]. This strategy is advantageous to obtain derivatives of a material in one step, and typically high-yielding reactions are employed, such as azide–alkyne and thiol–ene click reactions. The PSM conditions need to be carefully chosen not to jeopardize the integrity of the material and to prevent racemization of the chiral moiety being incorporated. The disadvantages of this strategy include the difficulties in determining the yield of the functionalization reactions, especially in the case of materials of high stability that cannot be digested to the respective building blocks for analysis. In addition, the spatial distribution of the chiral functionalities can be difficult to control in the case of non-quantitative reactions.
Although (iii) asymmetric synthesis is well established in organic synthesis [43], it has not been widely employed to date in the preparation of chiral porous organic frameworks. The attractive feature of this approach is that chirality is imparted by the catalyst material, and thus achiral starting materials can be used. However, determination of the degree of transfer of chiral information may not be trivial. In addition, finding the appropriate reaction conditions to ensure sufficient reversibility to yield reticular materials with long-range order could pose a challenge.
Another less commonly explored approach to chiral porous organic frameworks is (iv) chiral induction [44,45]. In this strategy, the chiral information is imparted by a compound that is present in the reaction medium but that does not become part of the final product, such as a chiral solvent or a chiral modulator, i.e., a compound that reversibly reacts with the building blocks. Although achiral compounds can be used as starting materials, the requirement for the building blocks is that they must have the ability to arrange in a chiral manner. Additionally, in this strategy, determining the degree to which chiral information has been incorporated into the resulting material is not trivial.
4. Chiroptical Responses
Chiroptical responses, those employing right and left circularly polarized light, have been extensively used in the last half a century for the structural characterization of chiral systems [73]. These spectroscopies feature larger sensitivity to the geometry of the system under study compared to the non-chiral spectroscopy counterparts, i.e., electronic circular dichroism (ECD) vs. ultraviolet/visible spectroscopy (UV/Vis). While UV/Vis is only proportional to the transition electric dipole moment, ECD is proportional to the dot product of the transition electric dipole moment and transition magnetic dipole moment (Figure 4). In the illustrated example, while the electron density displacement along the three chromophores present in the system (black arrows) features an overall transition electric dipole moment perpendicular to the macrocycle (gray arrow), this circulation of electron density around the cycle generates a transition magnetic dipole moment perpendicular to the cycle (orange arrow). However, the antiparallel or antiparallel alignment of transition electric and magnetic dipole moments depends on the chirality of the system (antiparallel/parallel in the illustrated (M,M,M)/(P,P,P)-enantiomer) [74]. This particularity enables not only the determination of the absolute configuration [75], the handedness of a system, but also the determination of the conformation [76] and even the characterization of host–guest complexes [77] and self-assembled systems [78].

Figure 4. Representation of transition electric dipole moment (gray arrows: total transition electric dipole moment; black arrows: the contribution from the different chromophores in the molecule) and transition magnetic dipole moment (red arrows) of the lowest electronic transition of a (M,M,M)-configured (left) and (P,P,P)-configured (right) cyclic spirobifluorene oligomer [74].
The dissymmetry factor, also referred to as the g-factor, is typically used to evaluate the chiroptical power of a system. In ECD, for instance, the g-factor is calculated as Δε/ε. For chiroptical responses arising from isolated systems such as discrete molecules in solution, the typical values of g-factor are in the range of 10−4–10−2, whereas for aggregated systems where the neighboring fragments are ordered in a chiral manner, the values can be 10−1 or higher [79]. While the chiroptical responses of most discrete molecules are evaluated in solution in transmittance mode, this approach can also be applied to insoluble systems by dispersing them in a solvent. An alternative for insoluble materials is the diffuse-reflectance mode on a suitable substrate.
This entry is adapted from the peer-reviewed paper 10.3390/catal13071042
This entry is offline, you can click here to edit this entry!