Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Neurodegenerative diseases are characterized by the progressive degeneration of nerve cells. Some neurodegenerative diseases such as Alzheimer’s and Parkinson’s are caused by disorders in the mitochondria, which are organelles present in the eukaryotic cells of animals, plants and fungi, and their function is to produce energy.
- mitochondrial neurodegenerative diseases
- mitochondrial ribosomal proteins
- MRPL44
- NAM9
- GEP3
1. Introduction
Luft et al. published in 1962 the first case of mitochondrial dysfunction in a 35-year-old woman affected by myopathy [1]. Since then, there has been a continuous advancement of research that is leading to the understanding, from a medical perspective, of the role of mitochondria in health, diseases and aging. Many seemingly unrelated pathologies, such as neuropathies, Alzheimer’s, Parkinson’s, myopathies, ataxia, cancer and others, share common underlying pathophysiological procedures. These mechanisms include the production of reactive oxygen species (ROS) and the damage they cause to the mitochondrial genome, leading to mitochondrial dysfunction [2,3,4]. As mitochondrial formation arises from the contribution of two distinct genomes, namely the nuclear DNA (nDNA) and the mitochondrial DNA (mtDNA) [5,6,7], genetic mutations in either the nDNA or the mtDNA, as well as genetic deletions in the mtDNA, can give rise to mitochondrial disease. These genetic mutations specifically result in defects in the mitochondrial oxidative phosphorylation system (OXPHOS) [8], which are the underlying cause of mitochondrial diseases [9,10,11]. Extensive research has been conducted on mutations occurring in nuclear genes associated with mitochondrial disorders, specifically focusing on their connection to diseases associated with premature aging [12,13] and POLG-related disorders that exhibit neurological symptoms [3,14,15]. Mitochondrial diseases represent the most common group of inherited metabolic dysfunction and are among the most prevalent types of inherited neurological disorders [16]. Mitochondrial disorders resulting from recurrent mutations in mtDNA manifest as shared syndromes across unrelated families and populations [2,17]. These disorders exhibit a wide range of clinical variations and can emerge at any age [16]. Given that mitochondria exist in all cells of the body except for red blood cells, the resulting clinical symptoms may manifest in specific organs independently but frequently involve multiple systems in organs that have high energy requirements, such as the brain, skeletal system, muscles and heart [16]. With reference to mutations in mtDNA or nDNA, from the point of view of clinical diagnosis, primary mitochondrial disease (PMD) and secondary mitochondrial dysfunction (SMD) are distinguished [18,19]. The primary differentiation between PMD and SMD lies in the fact that PMD genes either directly encode OXPHOS proteins or influence OXPHOS function by affecting the production of the complex machinery required for the OXPHOS process. On the other hand, SMD can be caused by genes that do not encode OXPHOS proteins or affect their production. SMD is often associated with various hereditary nonmitochondrial diseases [11,18,19]. There are several factors that determine the onset of mitochondrial dysfunction such as age, heredity factors, an unhealthy lifestyle or stress, environmental toxicity and more [7,16,20,21,22,23,24]. Among the mitochondrial diseases, the most widely known are neurodegenerative diseases, which are mainly caused by genetic factors and by environmental factors [7,24,25]. Neurodegenerative diseases are stress-inducing brain disorders characterized by behavioral, motor and cognitive impairments. Numerous studies conducted over the years have provided evidence that mitochondrial dysfunction serves as the underlying cause in the development of neurodegenerative diseases, which include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS) and Dementia [26,27,28,29]. These diseases arise from deficiencies in mitochondrial function, which is primarily regulated by over 1000 proteins encoded by both the mitochondrial and nuclear genomes [30]. Studies have shown that the majority, around 90%, of mitochondrial proteins are encoded by genes found in the cell nucleus. These proteins are synthesized by ribosomes in the cytosol and subsequently transported into mitochondria through a complex import machinery composed of multiple components [5,6,31,32,33,34]. These proteins have various functions attributed to them, including respiration, metabolite transport, protein translocation, redox homeostasis and other processes that are interconnected within complex and dynamic networks. The malfunctioning of these systems could lead to the development of diseases [35]. Despite mitochondria having their own genome, many nuclear-encoded mitochondrial ribosomal proteins (MRPs) are essential for proper organelle function. Mutations in Mrp genes, as indispensable components of the mitochondrial translation machinery, are detrimental to the OXPHOS system and associated with several neurodegenerative diseases in humans [36,37,38,39].
2. Mitochondrial Neurodegenerative Diseases
The human mitochondrial genome consists of a circular molecular structure comprising approximately 16,569 pairs of nucleotides (Figure 2). It contains a total of 24 mtDNA genes, including the small ribosomal RNA (12S rRNA) and large ribosomal RNA (16S rRNA) genes, as well as 22 transfer RNA (tRNA) genes necessary for translating the 13 respiratory chain proteins [19,41,42].
Studies conducted over the past forty years have highlighted the role of mitochondria in both normal brain function and the pathogenesis of diseases originating from them (Table 1) [43]. Mitochondrial diseases result from hereditary or spontaneous mutations in mtDNA or nDNA, leading to abnormalities in the functions of proteins or RNA molecules typically found within mitochondria [41,44,45]. Generally, the term “mitochondrial disease” describes a heterogeneous set of conditions caused by genetic defects in the assembly and/or function of OXPHOS proteins [45]. In fact, mitochondria play a fundamental role in energy production and are essential in numerous cellular processes [35,46]. Consequently, mitochondrial dysfunction often propagates and/or underlies many pathological states, including neurodegenerative diseases [47,48] and the aging process [49,50]. More recently, extensive research efforts have helped define some of the pathological mechanisms underlying neurodegenerative processes, such as the two most common neurodegenerative disorders: AD and PD.
Table 1. Mitochondrial diseases a.
| Mitochondrial Pathology | mtDNA Mutations | Clinical Syndromes | References |
|---|---|---|---|
| CPEO | single deletion | Loss of the muscle functions involved in eye and eyelid movement | [51,52] |
| KSS | single deletion | Neuromuscular disorder | [53,54] |
| PS | single deletion | It affects various parts of the body, especially bone marrow and the pancreas | [55,56] |
| Diabetes and deafness | single deletion | Hyperglycemia and reduction or absence of hearing ability | [57,58] |
| Encephalomyopathy | multiple deletions | Muscle weakness and pain, recurrent headaches, loss of appetite, vomiting and seizures | [59,60] |
| Recurrent myoglobinuria | multiple deletions | Metabolic disturbances that include hypokalemia, hypophosphatemia, hyponatremia, hypocalcemia and hypernatremia | [61,62] |
| SANDO | multiple deletions | Impaired coordination (ataxia), slurred speech (dysarthria) and weakness of the eye muscles (ophthalmoparesis) | [63,64] |
| LHON | point mutation | Progressive visual loss due to optic neuropathy | [65,66] |
| MELAS | point mutation | Disease primarily affecting the nervous system and muscles | [67,68] |
| NARP | point mutation | Neurogenic muscle weakness, sensory-motor neuropathy, ataxia and pigmentary retinopathy | [69,70] |
| MERRF | point mutation | Progressive myoclonus and seizures, cerebellar ataxia, myopathy, cardiac arrhythmia, sensorineural hearing loss, optic atrophy and dementia | [71,72] |
| CPEO | point mutation | Loss of the muscle functions involved in eye and eyelid movement | [19,73] |
| Leigh syndrome | point mutation | Neurological disorder involving elevated blood and/or cerebrospinal fluid levels of lactate, developmental retardation, hypotonia, followed by respiratory dysfunction, epileptic seizures, poor feeding and weakness | [74,75] |
| AD | nuclear gene mutation | Brain disorder that slowly destroys memory and thinking skills and, eventually, the ability to carry out the simplest tasks | [41,76] |
| PD | nuclear gene mutation | Combinations of motor problems—namely, bradykinesia, resting tremor, rigidity, flexed posture, “freezing,” and loss of postural reflexes | [76,77] |
| FRDA | nuclear gene mutation | Progressive ataxia, absent lower limb reflexes, upgoing plantar responses and peripheral sensory neuropathy. | [76,78] |
| HD | nuclear gene mutation | Disorder that causes nerve cells (neurons) in parts of the brain to gradually break down and die | [76,79] |
| ALS | nuclear gene mutation | Progressive nervous system disease that affects nerve cells in the brain and spinal cord, causing loss of muscle control | [76,80] |
| HSP | nuclear gene mutation | Disorder that causes the small blood vessels in skin, joints, intestines and kidneys to become inflamed and bleed | [76,81] |
| Aging | nuclear gene mutation | Accumulation of biological changes leading to functional decrease in the organism | [76,82] |
a Adapted with minor modification from Chinnery et al. [44]. CPEO = chronic progressive external ophthalmoplegia; KSS = Kearns-Sayre syndrome; PS = Pearson syndrome; SANDO = sensory ataxic neuropathy, dysarthria and ophthalmoparesis; LHON = Leber hereditary optic neuropathy; MELAS = mitochondrial encephalopathy, lactic acidosis and stroke-like episodes; NARP = neuropathy, ataxia and retinitis pigmentosa; MERRF = myoclonus epilepsy and ragged red fiber disease; AD = Alzheimer’s disease; PD = Parkinson’s disease; FRDA = Friedreich’s ataxia; HD = Huntington’s disease; ALS = amyotrophic lateral sclerosis; HSP = Henoch-Schönlein purpura.
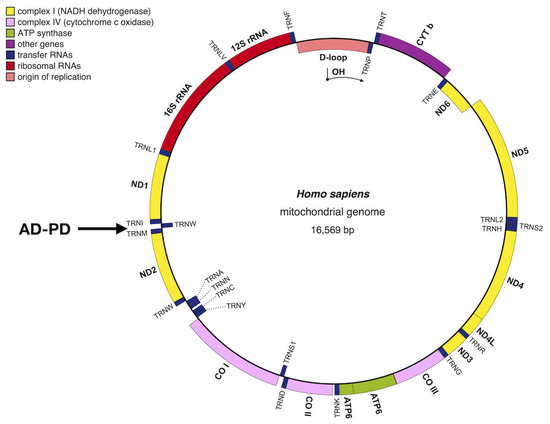
Figure 2. Schematic map of the human mitochondrial genome with related neurodegeneration diseases (adapted with minor modification from [2,19], CO: cytochrome oxidase (COX) subunit genes; CYTb = cytochrome b; ND = OXPHOS complex I subunit genes; D-loop = noncoding displacement loop or control region; AD-PD = neurodegenerative diseases: Alzheimer’s disease and Parkinson’s disease. Image generated with OGDRAW [83].
2.1. Alzheimer’s Disease in Brief
AD, a neurodegenerative condition linked to the aging process, is characterized by progressive cognitive and memory decline. It stands as the most prevalent neurodegenerative disorder, comprising around 60–70% of all dementia cases and impacting approximately 6% of individuals aged 65 and above (referred to as late-onset AD). Furthermore, a smaller subset of patients, around 2–10%, experience early-onset AD [41,84,85]. At the cellular level, AD is characterized by progressive and irreversible deterioration of neuronal structure and function in specific brain regions, including the hippocampus and neocortical areas, leading to cognitive dysfunction and dementia [86]. The main neuropathological features of the disease include the presence of extracellular β-amyloid plaques and intracellular neurofibrillary tangles formed by hyperphosphorylated tau proteins [87,88,89]. The causal relationship between the presence of these two hallmarks and AD is still not fully understood. It remains uncertain whether they are the primary cause of the disease or predominantly the outcome of a cascade of cellular events, including oxidative stress, mitochondrial dysfunction and apoptosis. Nonetheless, the exact mechanism by which these proteins damage neurons is still unknown [88,90]. Despite AD being the leading cause of dementia worldwide and the growing population of AD patients, no new therapies have received approval for over a decade [91,92].
2.2. Parkinson’s Disease in Brief
PD is the most common movement disorder and the second most frequent age-related neurodegenerative disorder in the world after AD [93,94]. It is a progressive and irreversible pathology characterized by significant neuronal loss in the pars compacta of the substantia nigra, leading to an alteration in dopaminergic transmission [95,96] and the presence of intracellular inclusions called Lewy bodies, which contain aggregates of α-synuclein [97,98]. The disease’s etiology is attributed to a combination of genetic and environmental factors [97]. In large populations, 5–10% of PD cases are attributed to known PD genes, indicating monogenic PD. However, 90 genetic risk variants collectively contribute only 16–36% to the genetic risk of nonmonogenic PD [94]. From a genetic standpoint, definitively characterized PD genes include autosomal dominant forms (SNCA, LRRK2 and VPS35) and autosomal recessive forms (PRKN, PINK1 and DJ1). Additionally, mutations in genes such as ATP13A2, DCTN1, DNAJC6, FBXO7, PLA2G6 and SYNJ1 have been associated with atypical or complex parkinsonism. Furthermore, recent studies have identified several other genes, including CHCHD2, LRP10, TMEM230, UQCRC1 and VPS13C, that are implicated in PD [77,99]. Besides core motor features such as bradykinesia (slowness of movement), rigidity and resting tremor, PD is also associated with a heterogeneous spectrum of nonmotor symptoms that significantly contribute to the overall disease burden of PD [100]. From a clinical perspective, there is no definitive method to diagnose PD in vivo, except for genetic testing in specific circumstances, which is limited to a few cases. Given the protracted timeline over which PD can advance, it imposes significant and far-reaching implications on patients, healthcare providers and society at large [94].
3. Mitochondrial Ribosomal Proteins Associated with Mitochondrial Neurodegenerative Diseases
Ribosomes are macromolecular machines with a universally conserved structure and function for protein synthesis. Within the cells of living organisms, mitochondria contain their own ribosomes, known as mitoribosomes, which are primarily responsible for synthesizing essential components of the oxidative phosphorylation machinery [41,101]. Mitoribosomes are located in the organelle matrix and are associated with the inner membrane to facilitate the cotranslational insertion of highly hydrophobic nascent polypeptides [38,102]. In mammals, mtDNA encodes 13 proteins that serve as crucial membrane components of the OXPHOS enzymatic complexes. The mammalian mitoribosome is a 55S ribonucleoprotein complex composed of a 39S large subunit (mt-LSU) containing 52 (MRPs), a 16S rRNA and a structural tRNA (tRNAVal in human cells), as well as a 28S small subunit (mt-SSU) with 30 MRPs and a 12S rRNA. In comparison to bacterial and eukaryotic cytoplasmic ribosomes, the 55S ribosomes contain a lower proportion of RNA, approximately 25–30% [103,104]. All MRPs are encoded in nDNA, synthesized using cytoplasmic ribosomes, and subsequently imported into the mitochondrial matrix. Once inside, they are assembled with subunit-specific RNAs encoded in mtDNA [41,104,105,106,107]. Dysfunctions in mitochondrial translation can lead to severe human diseases and may result from mutations in various components of the mitochondrial translation machinery, including tRNAs, aminoacyl-tRNA synthetases, translation factors and ribosomal components [2,108,109]. Furthermore, mutations or deficiencies in ribosome assembly proteins or other essential proteins can also contribute to mitochondrial diseases, as the mitochondrial ribosome is responsible for translating mRNAs for the 13 essential proteins of the OXPHOS system in affected tissues. Several MRP genes are located in loci associated with disorders related to impaired oxidative phosphorylation, such as Leigh syndrome, multiple mitochondrial dysfunctions and nonsyndromic hearing loss. Therefore, these diseases exhibit genetic heterogeneity and can manifest in a broad spectrum of clinical presentations [37,38,41,107,110,111]. The presence of numerous patients affected by mitochondrial disorders, characterized by multienzymatic OXPHOS defects of unknown genetic origin, suggests that there are still many genes involved in the biogenesis and function of the mitochondrial translation machinery that remain unidentified. Regarding alterations in ribosomal components associated with human diseases, only a limited number of mutations in mitoribosomal proteins have been reported so far [36,38,112]. In recent publications, the focus has been on the detection of pathological mutations in nuclear genes responsible for encoding mitochondrial ribosomal proteins. These findings emphasize the significance of their expression and involvement in mitochondrial translation, thus suggesting their potential role as contributing factors to genetic neurodegenerative diseases in humans [7,16,38,41,112,113,114].
4. Three Mitochondrial Ribosomal Proteins as Intermediate Stage in a Path Connected with Alzheimer’s and Parkinson’s
Neurodegenerative disorders are characterized by a gradual and progressive decline, specifically targeting interconnected neuronal systems based on anatomical or physiological relationships [115]. Mitochondria play a critical role in generating the majority of cellular ATP and are indispensable for ensuring optimal neuronal functioning. Dysfunction of mitochondria can give rise to PMDs and potentially contribute to the development of neurodegenerative conditions such as AD and PD. Mitochondria serve as crucial regulators of both cell survival and cell death, exerting a central influence on the aging process, and have been observed to interact with numerous specific proteins associated with genetic forms of neurodegenerative diseases [116]. These diseases include AD, PD, ALS and HD [115,117].
4.1. The Pathophysiology of Mitochondrial Diseases
The pathophysiology of mitochondrial diseases is complex and involves genetic mutations in both mtDNA and nDNA. Mitochondrial diseases are typically considered to be disorders caused by biochemical defects in the respiratory chain [117,118]. The regulatory role of nuclear genes in maintaining mitochondrial homeostasis and functionality is now included, along with defects in the lipid milieu, mitochondrial translation and mitochondrial fission and fusion [101,117,118]. Considerable progress has been made in the understanding of the molecular basis of mitochondrial diseases and their genetic etiology, particularly through next-generation sequencing/whole exome sequencing (NGS/WES) approaches. These approaches provide valuable information on the genes implicated in neurodegenerative disorders and could better define the impact of mitochondrial dysfunction on their pathogenesis [119]. As previously mentioned, mitochondrial diseases were caused as a consequence of mutations, whether inherited or spontaneous, in either mtDNA or nDNA. Furthermore, as previously discussed, human cells possess two distinct genomes and two protein synthesis systems. The first genome is the nuclear genome (nDNA), which consists of approximately 3 × 109 base pairs, and a second genome resides within a cytoplasmic organelle known as the mitochondrion (mt). Approximately 1500 nuclear gene products (around 3% of the total) are translated by cytoplasmic ribosomes and subsequently imported into the mitochondrion to perform their functions [120,121]. The cytoplasmic ribosome is composed of four rRNAs (28S, 18S, 5.8S and 5S) and 85 ribosomal proteins. The genes responsible for encoding these components have been mapped and investigated to a considerable extent [122]. Ribosomes play a crucial role as molecular machines, essential for all life on our planet. They decode information carried by messenger RNAs (mRNAs) and translate it into proteins [123]. Ribosome biogenesis is the crucial process in which all the components of ribosomes, such as ribosomal proteins (r-proteins) and rRNAs, come together to form the functional translation machinery. This process involves a large number of proteins, known as maturation factors and RNA factors (such as snoRNAs in cytosolic ribosome maturation) that aid in properly folding, assembling and modifying the different elements of the ribosome [124]. In mitochondria, the process of mitoribosome biogenesis is even more complex due to the need for coordination between two gene expression compartments [125]. Several mutations have been discovered in genes related to the cytoplasmic translation machinery, including ribosome components and numerous interacting factors. These mutations have been linked to various human diseases. On the contrary, genes from both the nuclear and mitochondrial genomes contribute to the formation of mitochondrial ribosomes, also known as mitoribosomes. These mitoribosomes play a crucial role in the synthesis of the oxidative phosphorylation machinery. These genes have been the subject of major research efforts in yeast and humans. Yeast serves as a model system for eukaryotic cell biology, while in humans, mitoribosomes have been implicated in human health [126]. The synthesis of ribosomal proteins occurs on cytoplasmic ribosomes, after which they are imported into the mitochondria. Interference with the synthesis of these proteins and other components of the mitochondrial translation system, such as mitochondrial tRNAs, either through deletion or mutation of the mitochondrial genes, is known to cause various mitochondrial diseases of varying severity. These diseases include myopathies and sensorineural disorders such as blindness and deafness [127]. By analogy, it can be expected that the loss or mutation of any of the 78 proteins required for the function of the mitochondrial ribosome [128] would also result in mitochondrial disease. Published research indicates that certain genes responsible for encoding MRPs are located within chromosomal loci that have already been associated with human disorders, including conditions like deafness, retinitis pigmentosa and Usher syndrome 1E [129]. A number of mutations that affect mitochondrial translation have been identified, including mutations in the core MRPs: MRPS16, MRPS22 and MRPL12 [130]. Additionally, mutations have been found in several accessory proteins, such as ObgH1 and C7orf30 [130]. It appears that mutations in MRPS16 and MRPS22 impact the stability of the mitoribosome [130], while MRPL12 is involved in the recruitment of elongation factors and the modulation of mitochondrial gene expression [129].
4.2. The Connection between Mitochondrial Dysfunctions and the Effects of Heavy Metals and Metalloid Oxyanions
Extensive scientific evidence has established a connection between mitochondrial dysfunctions, the harmful effects of heavy metals and metalloid oxyanions and systemic as well as neurodegenerative disorders [131,132]. Specifically, the toxicity of the metalloid tellurium (Te) has been implicated in the etiopathogenesis of various neurodegenerative disorders [132,133]. Accumulated experimental data indicate that tellurium not only exhibits a well-documented garlic-like odor but also exerts significant neurotoxic effects. In the past, Te has been employed as a valuable tool for studying the evolutionary origins of mitochondria in prokaryotes [134]. Mutations in these genes result in resistance to Te in the yeast S. cerevisiae. Additionally, previous publications have associated the toxic effects of Te with the development of neurodegenerative diseases like AD and PD [132,133]. The recent work by Pontieri et al. [40] represents a significant advancement in scientific knowledge regarding the study of the three MRPs involved in potassium Te resistance in S. cerevisiae. It sheds light on their potential role in neurodegenerative dysfunctions such as AD or PD, thus providing valuable insights for further investigation.
This entry is adapted from the peer-reviewed paper 10.3390/biology12070972
This entry is offline, you can click here to edit this entry!